【论文阅读】SIMBA: single-cell embedding along with features(1)

代码地址:https://github.com/pinellolab/simba
摘要
当前大多数单细胞分析流程仅限于细胞嵌入,并且严重依赖聚类方法,而缺乏显式建模不同特征类型之间相互作用的能力。此外,这些方法往往针对特定任务进行定制,因为不同的单细胞问题通常以不同方式被提出。
为了解决这些不足,我们提出了 SIMBA ——一种图嵌入方法,它能够将单细胞及其定义特征(如基因、染色质可及区域和 DNA 序列)共同嵌入到一个共享的潜在空间中。通过利用细胞与特征的协同嵌入,SIMBA 可以支持研究细胞异质性、无聚类的标记物发现、基因调控推断、批次效应去除以及多组学数据整合。
我们展示了 SIMBA 如何提供一个统一的框架,使得多样化的单细胞问题能够在一致的方式下被表述,从而简化新分析方法的开发和向新单细胞模态的扩展。SIMBA 已实现为一个全面的 Python 库(https://simba-bio.readthedocs.io)。
引言
单细胞多组学背景与 SIMBA 方法
近年来,单细胞组学技术的进展使得对细胞层面的独立和联合检测成为可能。单细胞多组学技术的出现,使得我们能够同时测量多种细胞层级的信息,包括基因组学、表观基因组学、转录组学和蛋白质组学。这类检测极大提升了我们理解细胞状态以及发育和疾病背后分子机制的能力。尽管这些技术潜力巨大,但如何在计算层面充分发挥其能力仍面临挑战。
目前已有许多单细胞计算方法被开发用于单一模态的分析(如 单细胞 RNA 测序(scRNA-seq) 或 单细胞 ATAC 测序(scATAC-seq))¹–⁴。这类方法通常遵循一个标准流程,包括特征选择、降维、聚类以及差异特征检测。这些以“聚类为中心”的分析方法高度依赖于聚类结果的准确性,以便发现有意义的标记特征。然而,聚类结果在用户定义的聚类分辨率(即簇的数量)和选择的聚类算法之间可能差异很大,从而导致生物学注释的不一致甚至错误⁵。
尽管已有一些初步尝试发展无聚类方法来发现信息性基因,这些方法主要集中在从 scRNA-seq 数据中提取基因特征⁶,⁷,或在实验条件之间识别扰动⁸,因此仍局限于单模态和单任务分析。
此外,也有一些计算方法被提出用于多批次和跨模态分析,例如:
多模态分析:在同一细胞中同时测量不同参数⁹;
批次校正:在不同批次中测量相同参数¹⁰–¹²;
多组学整合:在不同细胞中测量不同参数¹¹,¹²。
然而,由于任务定义方式不同,这些任务通常需要开发新的方法。此外,大多数现有方法无法直接利用多种细胞特征之间的关系。而且,这些用于识别标记特征的方法依赖于聚类,因此受限于聚类结果。另外,目前最先进的批次校正和多组学整合方法¹⁰–¹²,并不能直接在整合后的空间中识别标记特征,而是必须先在每个批次或模态的原始未校正空间中分别检测标记特征,再进行合并,这容易导致不同批次或模态之间的解释不一致。
SIMBA 方法
为克服这些局限性,我们提出了 SIMBA(single-cell embedding along with features),一种通用的单细胞嵌入方法。SIMBA 将细胞及其特征(如基因、染色质开放区域、DNA 序列等)共同嵌入到一个共享的潜在空间中,从而能够以统一的方式执行多种任务。
与现有需要先对细胞进行特征化的方法不同,SIMBA 直接将细胞—特征或特征—特征之间的关系编码为一个大型多关系(即包含多类节点和边)的图。在每个任务中,SIMBA 构建这样一个图:细胞和特征作为节点,二者之间的关系作为边。一旦图被构建,SIMBA 就会应用源自社交网络技术的多关系图嵌入算法¹³,¹⁴,以及基于 Softmax 的转换,将节点或实体嵌入到一个共享的低维空间中,使得细胞和特征可以根据距离进行分析。
因此,SIMBA 嵌入空间中包含的细胞与所有特征可以被视为一个信息丰富的数据库。根据任务需求,可以在这个“SIMBA 数据库”上定义生物学查询,分别以单细胞或单特征为单位来考察其邻近实体(方法部分)。例如:
对某个细胞的邻近特征进行查询,可以用于识别标记特征(如标记基因或染色质峰),或研究特征间的相互作用(如基因-峰关系);
对某个特征的邻近细胞进行查询,则可用于细胞注释。
这与最近提出的单细胞嵌入方法有本质区别(补充说明 1 与补充表 1)。
应用与优势
SIMBA 在一个统一框架下能够解决多种单细胞任务,包括:
降维分析;
无聚类的标记物检测;
多模态分析;
批次校正与多组学整合。
通过对输入图的灵活构建,SIMBA 可以轻松适应这些不同任务。我们在多个 scRNA-seq、scATAC-seq 及双组学数据集上对 SIMBA 进行了广泛测试,结果显示其性能优于或至少不逊于为各任务单独开发的当前最先进方法。
重要的是,我们开发了一个可扩展、全面的 Python 包,支持图构建、基于 PyTorch 的图嵌入训练以及训练后的分析之间的无缝交互。SIMBA 是一个自包含的框架,但同时也兼容常用的单细胞分析工具,如 Scanpy²。
方法
SIMBA 概述
SIMBA 是一种单细胞嵌入方法,支持 单模态 和 多模态分析。它利用最新的 图嵌入技术¹³,¹⁴,将细胞与基因组特征嵌入到共享的潜在空间中。与现有主要专注于学习细胞状态的方法不同,SIMBA 将细胞和特征都视为同一图中的节点,从而通过统一的流程解决多种单细胞任务。
值得注意的是,SIMBA 引入了若干关键步骤,包括 Softmax 转换、权重衰减(控制过拟合)以及 实体类型约束,以生成细胞和特征的可比较嵌入(协同嵌入),并应对单细胞数据中的独特挑战。
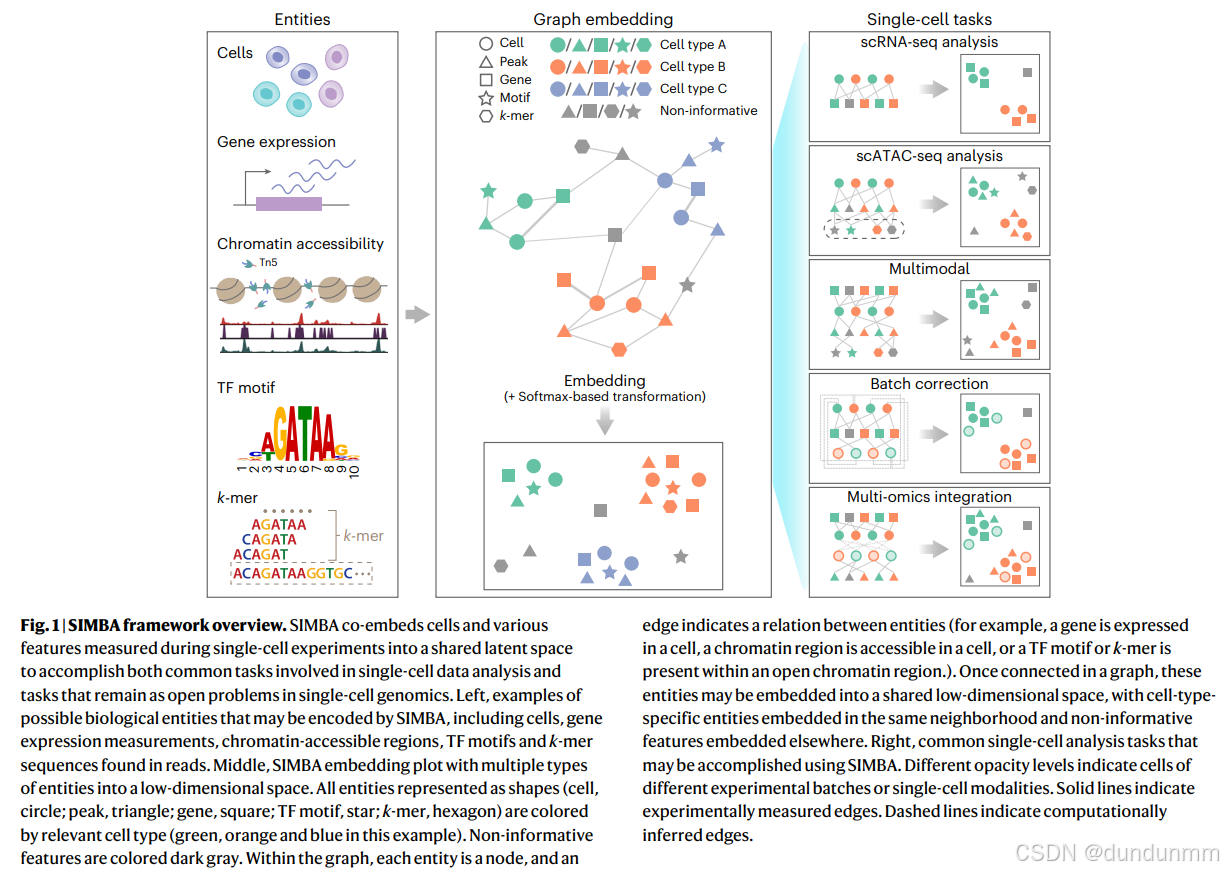
在 SIMBA 中,不同类型的实体(如细胞、基因、开放染色质区域〔peaks 或 bins〕、转录因子(TF)motifs 以及 k-mers〔长度为 k 的短序列〕)被编码进一个统一的图中(图 1 和方法部分)。其中,每个节点代表一个独立的实体,边表示实体之间的关系。
实验测量边:例如,若一个基因在某细胞中表达,就在该基因与细胞之间建立一条边,边的权重由基因表达水平决定。类似地,如果某个染色质区域在该细胞中开放,则在该细胞和该染色质区域之间建立一条边。
特征间的边:用于捕捉和建模潜在的调控机制。例如,若一个峰(chromatin peak)的序列包含某个 TF motif 或 k-mer,则在该峰和该 motif(或 k-mer)之间建立边,表示该 TF 可能结合于该调控区。
计算推断边:无法直接测量的关系可通过计算推断得出,例如不同批次或不同模态细胞之间的边,表示其功能或结构上的相似性。
图 1 总结了这些边的关系语义:
(cell–gene):细胞表达某基因;
(cell–peak):细胞具有一个开放的染色质区域;
(peak–TF motif):峰序列包含某 TF 的结合位点;
(peak–k-mer):峰序列包含某特定 k-mer;
(cell–cell):不同批次或模态的细胞在功能或结构上相似。
一旦输入图构建完成,SIMBA 会利用 无监督图嵌入方法 计算节点的低维表示,基于 PyTorch-BigGraph 框架¹⁴,该框架可扩展至数百万级细胞。最终得到的细胞与特征的联合嵌入不仅能重构细胞异质性,还能在不依赖聚类的情况下发现决定性特征,从而区分 细胞类型特异性(信息性)特征 与 非特异性(非信息性)特征。
在 SIMBA 嵌入空间中,节点之间的接近程度反映了边的存在概率,这一概率既表征了特征的重要性,也揭示了特征之间的相互作用(见方法部分)。因此,细胞类型特异性特征(如标记基因、顺式调控元件)可以通过两种方式被发现:
已知细胞标签时:通过生物学查询寻找邻近特征;
未知标签时:通过计算特征与所有细胞之间边概率的不平衡性(如 Gini 指数)来识别。
更为重要的是,图构建的高度灵活性使 SIMBA 能够适用于多种单细胞任务。接下来,我们展示 SIMBA 在以下常见任务中的应用:
scRNA-seq 分析
scATAC-seq 分析
多模态分析
批次校正
多组学整合(见图 1)。
单细胞数据预处理
单细胞 RNA-seq
在 scRNA-seq 中,表达于少于三个细胞的基因会被过滤。原始计数经过文库大小标准化(library-size normalization),随后进行对数转换。可选地,可以执行可变基因选择¹²(SIMBA 实现了一个基于 Python 的版本,灵感来源于 Scanpy²),以去除非信息性基因并加快训练过程。值得注意的是,当仅输入可变基因时,得到的细胞嵌入与未筛选时并无显著差异,但非可变基因不会被编码进图中,因此不会生成对应的嵌入。
单细胞 ATAC-seq
在 scATAC-seq 中,出现在少于三个细胞中的峰(peaks)会被过滤。可选地,SIMBA 实现了一种可扩展的基于截断 SVD的方法来选择可变峰,作为过滤非信息性峰的预处理步骤,并加快训练过程。
首先选择前 k 个主成分(PCs),k 的选择基于方差比例的“肘部图”。
接着,在每个 PC 上,利用“kneed”算法⁴¹自动检测负载系数的拐点,选择对应的峰。
最后,将每个 PC 选出的峰合并,作为“可变峰”。
与 scRNA-seq 的观察类似,可变峰选择对最终嵌入的影响可以忽略不计,但该步骤在实践中能明显缩短训练时间。
此外,k-mer 与 motif 扫描通过 R 包 Biostrings 与 motifmatchr 并结合 JASPAR2020 数据库⁴² 进行。SIMBA 提供了一个便捷的 R 脚本 scan_for_kmers_motifs.R,可将峰列表(bed 文件格式)转换为稀疏的 peak-by-k-mer/motif 矩阵,并存储为 HDF5 格式。
图构建(五种场景)
scRNA-seq 分析
在构建细胞-基因图时,若某基因在某细胞中表达,则在两者之间建立一条边。为区分边的强度,提出了分箱(binning)方法,将基因表达值划分为多个等级,同时保留原始分布。不同表达水平由不同的关系类型编码。
具体而言:
归一化基因表达矩阵中的非零值先通过基于 k-means 聚类的过程近似分布;
连续非零值被分箱为 n 个区间(默认 n=5),每个区间的宽度由一维 k-means 聚类确定;
最终矩阵被转换为离散矩阵,其中 1…n 表示 n 个基因表达水平,零值保持不变。
最终图的构建方式:细胞和基因为节点,不同水平的基因表达作为边的 n 种权重,范围 1.0–5.0(步长 5/n),表达水平越高,对嵌入的影响越强。实践表明,增加分箱数量能使离散分布更接近原始分布,但对嵌入结果影响很小。SIMBA 中通过 si.tl.discretize() 函数实现该步骤。
除了分箱编码,SIMBA 还支持直接将基因表达值作为边权重。补充图 S25 显示,无论是采用原始表达值、离散值还是分箱权重,最终嵌入结果均相似,这说明分箱方法能有效捕捉生物学信息,且 SIMBA 对分箱过程具有鲁棒性。该功能通过 si.tl.gen_graph(add_edge_weights=True) 实现。
scATAC-seq 分析
peak-by-cell 矩阵被二值化:
“1” 表示该细胞中存在至少一个读取落在某个峰内;
“0” 表示不存在。
构建的图由细胞和峰作为节点,边表示某细胞是否包含该峰,单一关系权重设为 1.0。若 DNA 序列特征可用,可通过 k-mer 和 转录因子(TF)motif 扩展该图:
先二值化 peak-by-k-mer/motif 矩阵;
再在原始 peak-cell 图基础上扩展,新增 k-mer 与 motif 节点,并通过边连接到相应的峰节点。
关系权重设置为:k-mer 与峰 = 0.02,motif 与峰 = 0.2。k-mer 和 motif 可根据具体任务独立或联合作为输入。
多模态分析
将上述 scRNA-seq 与 scATAC-seq 的图构建策略结合,生成多组学图。
批次校正
每个批次的图按 scRNA-seq 的方法构建,不同批次细胞之间的边则通过截断随机化 SVD推断,以连接不同批次的分离图。
具体来说:
给定两个基因表达矩阵 X₁(n₁×m) 和 X₂(n₂×m),其中 n₁、n₂ 为细胞数,m 为共享特征数(即可变基因);
计算矩阵 X = X₁ × X₂ᵀ;
对 X 执行截断随机化 SVD:
X≈U×Σ×VTX其中 U (n₁×d)、Σ(d×d)、V(n₂×d),默认 d=20;
U 与 V 进一步进行 L2 归一化;
对 U 中的每个细胞,寻找其在 V 中的 k 个最近邻,反之亦然(默认 k=20);
最终,仅保留 U 与 V 之间的互为最近邻作为跨批次的推断边(图 5a 虚线所示)。
该过程在 SIMBA 中由 si.tl.infer_edges() 实现。对于多个批次,SIMBA 可灵活地在任意数据集对之间推断边,但实践中,通常选取规模最大或细胞类型最完整的数据集作为参照,与其他批次建立连接。
多组学整合
scRNA-seq 与 scATAC-seq 图构建
scRNA-seq 和 scATAC-seq 的图分别按照“单细胞 RNA-seq 分析”和“单细胞 ATAC-seq 分析”的步骤进行构建。为推断两者之间细胞的边,首先计算 scATAC-seq 的基因活性得分³:
对于每个基因,考虑其转录起始位点(TSS)上下游 100 kb 范围内的峰;
与基因体重叠或位于基因体上游 5 kb 内的峰,权重设为 1.0;
其他峰则根据与 TSS 的距离,按指数衰减函数 e−distance/5000赋权;
每个基因的得分为加权峰的总和,并缩放至对应的基因大小。
这些步骤由 SIMBA 中的 si.tl.gene_scores() 函数实现,且内置了常用参考基因组(hg19、hg38、mm9、mm10)的注释。获得基因得分后,按照“批次校正”中的方法,使用 si.tl.infer_edges() 推断 scRNA-seq 与 scATAC-seq 细胞之间的边。完整图的生成由 si.tl.gen_graph() 函数实现。
图嵌入与类型约束
在构建了生物实体之间的多关系图后,SIMBA 借鉴知识图谱和推荐系统中的图嵌入技术,为这些实体学习无监督表示。
输入是一个有向图 G=(V,E),其中 V 为实体集合(节点),E 为边集合,每条边 e=(u,v) 表示从源实体 u 到目标实体 v 的关系。假设每个实体有已知的类型(如细胞或峰)。
图嵌入方法为每个节点 v 学习一个 D 维向量(实验中取 D=50),并通过随机梯度下降优化链路预测目标函数。记总嵌入矩阵为 Θ∈R∣V∣×D,单个实体 v 的嵌入为 θv。对于边 e=(u,v),得分定义为 se=θu⋅θv,优化的多分类对数损失为:
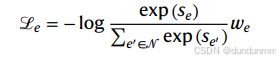
其中 N是通过破坏原始边构造的负样本集合⁴³,we 为边权重(默认为关系权重,也可在同一关系类型内随边变化)。例如,细胞-基因边可以被编码为单一关系,但边权重则表示归一化的基因表达水平(参见“单细胞 RNA-seq 分析”)。优化目标是最大化真实边的得分、最小化负边的得分。
负样本采样:通过随机替换边的源或目标实体生成。但由于本图仅允许特定实体类型间的边,优化时仅保留符合类型约束的候选负样本⁴⁴。例如,对细胞-峰边,只在细胞与峰之间采样负例,而不考虑无效的峰-峰边。这一点十分关键,否则嵌入会被迫学习大量无关约束。
在节点度分布差异较大的图中,已有研究表明,按节点度的函数比例进行采样有利于学习更具信息性的嵌入⁴⁵。因此,在训练中,每条边会生成 200 个负例:100 个从同类型节点中均匀采样,100 个按节点度加权采样。
正则化与优化:与许多机器学习方法类似,在低数据量(边数与参数数比值低)的情况下,图嵌入易发生过拟合。我们通过在嵌入上添加 L2 正则项解决该问题:
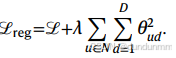
其中 λ=wd×wdinterval。默认情况下,权重衰减参数 wd自动计算为 C/Ne(Ne 为训练样本数,即总边数,C 为常数),wdinterval在所有实验中均设为 50。
SIMBA 使用 PyTorch-BigGraph 框架实现高效的多关系图嵌入训练,可扩展到数百万甚至数十亿节点¹⁴。在 130 万细胞的实验中,仅用 12 个 CPU 核心即可在约 1.5 小时内完成训练,无需 GPU。
学到的嵌入的两个特性
一阶相似性(first-order similarity)
对于两类实体 T₁ 和 T₂,若存在关系,则高可能性的边应具有更高的点积。例如,对于任一 u∈T₁,预测从 u 到 T₂ 的边分布为: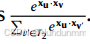
二阶相似性(second-order similarity)
在同一类型内,若两个实体具有相似的上下文(即相似的边分布),则其嵌入应相似。因此,每类实体的嵌入提供了一个低秩潜在空间,编码了该类实体的边分布相似性。
模型训练过程中的评估
在 PyTorch-BigGraph 训练过程中,会保留一小部分边(默认评估比例为 5%),用于监控过拟合并评估最终模型。针对保留的边集计算五个指标:
MRR(平均倒数排名,所有正例排名倒数的平均值);
R1(正例排名优于所有负例的比例,即排名=1 的比例);
R10(正例在负例中排名前 10 的比例);
R50(正例在负例中排名前 50 的比例);
AUC(曲线下面积)。
默认情况下,SIMBA 会显示 MRR,以及训练损失和验证损失,其他指标在软件包中也可用(补充图 S1a)。验证损失与这些指标的学习曲线可用于判断训练何时完成。训练损失与验证损失的相对值结合评估指标,可以帮助识别训练问题(欠拟合 vs. 过拟合),并指导调整超参数(如权重衰减、嵌入维度、训练轮数)。例如,在补充图 S1a 中,一旦验证损失进入平台期,训练即可停止。但对于大多数数据集,默认参数已足够(补充说明 9)。
Softmax 转换
PyTorch-BigGraph 训练可生成所有实体(节点)的初始嵌入。然而,不同类型的实体(如细胞与峰、不同批次或不同模态的细胞)具有不同的边分布,因此可能位于潜在空间的不同流形上。为使不同类型实体的嵌入具有可比性,我们使用 Softmax 函数对特征嵌入进行转换,基于细胞(参考)与特征(查询)之间的一阶相似性。
在批次校正或多组学整合中,Softmax 转换同样基于不同批次或模态的细胞之间的一阶相似性。
设细胞(参考)的初始嵌入为 (vc1,…,vcn),特征(查询)的嵌入为 (vf1,…,vfm),则模型估计边 (ci,fj)的概率为:
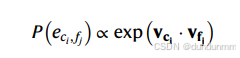
若随机采样从特征 fjf_j 到细胞的边,则其分布为:
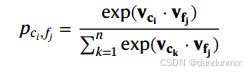
这定义了特征 fjf_j 与所有细胞的 Softmax 权重。新的特征嵌入可表示为加权的细胞嵌入的线性组合:
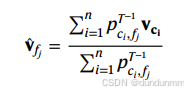
其中 T 为温度超参数,控制权重分布的“尖锐度”:
T=1 时,按概率加权;
T→0 时,特征嵌入取最近邻细胞嵌入;
T→∞ 时,取均匀分布,即参考嵌入的平均值。
在所有分析中,T 设为 0.5。这些步骤在 SIMBA 中由 si.tl.embed() 实现。
细胞类型特异性的评估指标
SIMBA 会为每个特征计算其分配到各个细胞的概率分布(点积表示),据此可以用四个指标从不同角度评估细胞类型特异性:
Max score⁴⁶:前 50 个细胞的归一化概率的平均值,衡量对特定细胞类型分配的置信度。值越高,特异性越强。
Gini 指数⁴⁶:基于 Softmax 分布计算,评估与完全均匀分布的偏离程度。值越高,表示分布越不均匀,即更具细胞类型特异性。
标准差(s.d.):度量概率分布的离散程度,值越高表示特异性越强。
熵(Entropy):度量分布的信息量,值越低表示分布集中于某一类细胞,因而特异性更高。
这四个指标通常结果一致。默认情况下,SIMBA 绘图将 Gini 指数 与 Max score 联合展示。
公式定义:
Max 值:前 k 个细胞相似度的平均,默认 k=50。
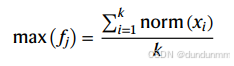
其中 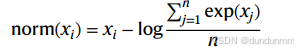 。
。
Gini 指数:
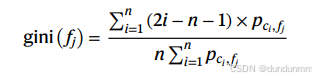
标准差:
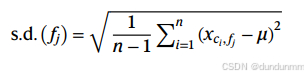
熵:
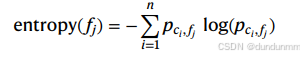
显著性评估
这些指标的显著性可通过与“空(null)特征节点”的对照分布比较来计算。空节点通过在保持节点度分布一致的前提下随机打乱边构建,从而保留节点属性偏差但破坏图的连接性。
例如:
在 scRNA-seq 中,将真实基因与细胞节点和“空基因节点”一起构建图,空基因的边随机打乱;
在间接特征(如 TF motifs、k-mers)的情况下,打乱它们与峰之间的边。
训练时,空节点的损失不会影响真实节点嵌入(通过 fix 操作实现)。默认情况下,空节点数量为基因数的 20 倍(scRNA-seq)或峰数的 5 倍(scATAC-seq、SHARE-seq)。最终,基于空分布计算 P 值,并使用 Benjamini–Hochberg 方法校正 FDR。
SIMBA 空间中的实体查询
信息丰富的 SIMBA 嵌入空间可作为包含细胞和特征的数据库。要查询某个细胞或特征的邻近实体,我们首先基于 SIMBA 嵌入构建所有实体的 k-d tree,并使用欧式距离进行最近邻搜索。SIMBA 支持两种查询方式:
k 最近邻(KNN)搜索;
指定半径范围内的最近邻搜索。
同时,SIMBA 提供限制搜索实体类型的选项,这在某类实体数量远大于其他类型时非常有用。例如,在查询某个细胞的最近邻特征时,可能返回的全是峰(peaks),但研究者关心的却是基因。在这种情况下,SIMBA 允许用户添加“过滤条件”,以确保最近邻搜索仅在指定类型实体内进行。该功能由 st.tl.query() 实现,可视化由 st.pl.query() 实现。
主调控因子的识别
在 SIMBA 中,我们将 主调控因子(master regulator) 定义为其 转录因子(TF)motif 和 TF 基因 都具有细胞类型特异性,因为有效的基因调控通常涉及 TF 的表达及其结合位点的可及性。因此,TF motif 与 TF 基因应在共享潜在空间中彼此接近。
识别流程:
计算每个 TF motif 与所有基因的距离(默认欧式距离);
得出该 TF 基因在所有基因中的排序;
结合 SIMBA 的特异性指标(默认使用 Max 值和 Gini 指数)评估该 TF 基因与 motif 对的细胞类型特异性;
对所有 TF 重复以上步骤,并过滤掉特异性较低的 TF,最终根据 TF 基因排序筛选出主调控因子。
该方法在 st.tl.find_master_regulators() 中实现。
TF 靶基因的识别
为推断某个主调控因子的靶基因,我们提出以下假设:
靶基因应同时靠近 TF motif 与 TF 基因;
靶基因附近的可及区域(peaks)应靠近 TF motif 与 TF 基因,表明靶基因的调控元件可及性与 TF 表达和 motif 可及性高度相关。
识别流程(图 4e):
对某主调控因子的 motif 和基因,分别搜索其 200 个最近邻基因,并取并集作为候选靶基因集;
筛选:候选靶基因的 TSS 上下游 100 kb 内的开放区域必须包含该 TF motif;
对每个候选靶基因,计算四类距离:
候选靶基因与 TF 基因;
候选靶基因与 TF motif;
候选靶基因附近峰与 TF motif;
候选靶基因附近峰与 TF 基因。
距离转换为排序(默认欧式距离),以便不同 TF 之间结果可比;
最终筛选条件:
至少一个峰在前 1,000 个最近邻内;
候选靶基因的平均排序在前 5,000 内。
该方法由 st.tl.find_target_genes() 实现。
scATAC-seq 方法基准测试
为比较 SIMBA 与其他 scATAC-seq 计算方法(SnapATAC⁴、Cusanovich⁴⁷、cisTopic⁴⁸),我们使用了 Chen 等人提出的基准框架¹⁶(补充表 2)。该框架依据区分细胞类型的能力评估方法性能。
聚类算法:k-means、层次聚类、Louvain;
数据集:模拟骨髓数据、Buenrostro 等数据¹⁷、sci-ATAC-seq 子集;
评价指标:ARI(调整兰德指数)、AMI(调整互信息)、Homogeneity(同质性)。
ARI 衡量预测聚类与真实标签的一致性;
AMI 衡量变量间的互依赖性;
Homogeneity 衡量聚类是否将同一类别的样本归入同一簇。
对于没有真实标签的 10x PBMCs 数据集,使用 RAGI(残差平均 Gini 指数) 作为聚类评价指标。RAGI 衡量标记基因在簇中的专属性,相比之下 housekeeping 基因应表现为低特异性。最终通过标记基因与 housekeeping 基因的平均 Gini 值差异来计算 RAGI,数值越高说明生物学上簇的分离越好。
单细胞批次校正方法基准测试
我们比较了 SIMBA 与 Seurat3¹²、LIGER¹¹、Harmony¹⁰ 的批次校正性能,使用小鼠图谱和人类胰腺两个基准数据集(补充表 2)。这些方法的参数与之前的基准研究²⁸保持一致。
评估指标:
ASW(平均轮廓宽度);
ARI;
LISI(局部逆 Simpson 指数)。
这些指标分别针对批次与细胞类型计算。为了公平比较,仅保留所有批次中都存在的细胞类型。所有方法均使用 50 个维度。
单细胞多组学整合方法基准测试
为评估模态整合性能,我们使用了 SHARE-seq 小鼠皮肤数据集和 10x PBMCs 数据集,手动拆分为 scRNA-seq 与 scATAC-seq 两部分。
Seurat3 与 LIGER:按官方文档进行预处理与参数设置;
对 SHARE-seq 数据集,LIGER 的参数
lambda设为 30,ref_dataset设为 scATAC-seq,以获得更佳对齐效果;Raw 结果:scATAC-seq 的活性矩阵由 Seurat3 构建,scRNA-seq 计数矩阵与活性矩阵的前 20 个主成分用于比较。
评估指标包括:
锚点距离;
锚点距离排名;
轮廓系数;
聚类一致性。
结论
见下一篇。