生信分析自学攻略 | R语言数据类型和数据结构
在前面两篇文章中,我们已经成功搭建了R和RStudio这一强大的生信分析平台。然而,工具再好,若不懂得如何“放置”和“理解”你的数据,一切都将寸步难行。今天,我们将学习R语言最重要的部分——数据类型(Data Types) 和 数据结构(Data Structures)。
测序数据一般包括基因名、基因ID、表达量、样本信息、分组标签……如果这些数据杂乱无章,你如何能从中挖掘出有意义的生物学结论?R语言通过其精妙的数据类型和丰富的数据结构,为我们提供了将“数据乱码”整理成“有序信息”的强大能力。
本篇,我们不仅仅停留在概念讲解,而是将这些基础知识与真实的生信分析场景紧密结合,在解决问题的过程中,体会数据管理的艺术。
R语言的数据类型:数据的“本质属性”
在R中,每个数据都有其特定的数据类型,这决定了R如何存储和处理它。理解数据类型是后续构建数据结构的基础。最常见的几种数据类型包括:
- 数值型(Numeric):用于存储数字,包括整数(Integer)和双精度浮点数(Double)。
应用场景: 基因表达量(如TPM、FPKM、Counts)、样本年龄、实验重复次数等。
示例:c(100.5, 230, 0.01) - 字符型(Character):用于存储文本信息。
应用场景: 基因ID(如"TP53")、样本名称(如"Sample_WT_Rep1")、通路名称、GO富集结果的描述等。
示例:c("GeneA", "GeneB", "GeneC") - 逻辑型(Logical):只包含 TRUE (真) 或 FALSE (假) 两种值,常用于条件判断。
应用场景: 判断基因是否上调/下调、样本是否属于疾病组、质控是否通过等。
示例:c(TRUE, FALSE, TRUE) - 因子型(Factor):特殊的一种字符型,用于存储有限个预定义类别的数据,在统计建模中非常重要。R会将其视为分类变量。
应用场景: 实验分组(如“Control” vs “Treatment”)、性别(“Male” vs “Female”)、细胞类型(“B_cell”, “T_cell”, “Macrophage”)等。
示例:factor(c("Control", "Treatment", "Control"))
为何重要: 在进行差异表达分析或回归分析时,R会自动识别因子变量,并将其正确地纳入统计模型,处理起来比普通字符型更加高效和准确。
R语言的数据结构:数据的“组织形式”
数据类型给数据“安上了标签”,数据结构就是将这些带标签的数据梳理成有规律的数据集,便于函数处理,进而发现生物学规律。
向量(Vector):一维数据的序列
定义: 向量是R中最基本的数据结构,用于存储同一种数据类型的元素序列。
- 应用场景:
单个基因在不同样本中的表达量。
一系列样本的测序深度。
一组基因的差异表达P值。
单个样本的质控指标(如线粒体基因比例)。 - 操作与理解:
使用c()函数创建向量。
通过索引([])访问和修改元素。
进行向量化的运算(无需循环,R会自动对每个元素执行操作,效率高)。
【实战演练:批量计算P值校正】
假设我们通过差异表达分析得到了10个基因的原始P值,现在需要进行FDR(False Discovery Rate)校正。
# 1. 创建一个数值型向量,存储原始P值
raw_p_values <- c(0.001, 0.045, 0.12, 0.0005, 0.08, 0.003, 0.5, 0.06, 0.009, 0.02)
print("原始P值:")
print(raw_p_values)# 2. 使用p.adjust函数进行FDR校正(这是一个典型的向量化操作)
# "fdr" 是校正方法,也可以是 "bonferroni", "BH" 等
adjusted_p_values <- p.adjust(raw_p_values, method = "fdr")
print("FDR校正后的P值:")
print(adjusted_p_values)# 思考:如果raw_p_values中混入了字符型数据,例如 "NA",p.adjust函数还能正常工作吗?为什么?

矩阵(Matrix):二维的同类型数据表
定义: 矩阵是二维的数组,所有元素必须是同一种数据类型。它有行和列,非常适合存储表格型数据。
- 应用场景:
基因表达矩阵: 行是基因,列是样本,矩阵中的值是基因的表达量(数值型)。这是转录组和单细胞分析中最核心的数据结构之一。
距离矩阵、相似性矩阵等。 - 操作与理解:
使用matrix()函数创建。
通过dim()查看维度,nrow()获取行数,ncol()获取列数。
通过matrix[row_index, col_index]进行索引。
【实战演练:主成分分析(PCA)的数据准备】
在转录组或单细胞分析中,PCA是一种常用的降维和可视化方法,用于查看样本间的整体关系或细胞群的异质性。PCA通常需要一个基因表达矩阵作为输入。
场景: 假设我们有3个样本(SampleA, SampleB, SampleC),和4个基因(Gene1-Gene4)的表达量。
# 1. 创建一个基因表达矩阵
# 数据顺序:按列填充,即先填SampleA的Gene1-4,再填SampleB的Gene1-4...
expression_data <- matrix(c(10, 15, 20, 25, # SampleA12, 18, 22, 28, # SampleB30, 25, 18, 12), # SampleCnrow = 4, ncol = 3,byrow = FALSE, # 默认按列填充,即先填满第一列再第二列dimnames = list(c("Gene1", "Gene2", "Gene3", "Gene4"), # 行名:基因c("SampleA", "SampleB", "SampleC") # 列名:样本)
)
print("基因表达矩阵:")
print(expression_data)# 2. 对矩阵进行转置,使行是样本,列是基因(PCA通常要求样本在行)
# t() 函数用于转置矩阵
transposed_expression_data <- t(expression_data)
print("转置后的矩阵(样本在行,基因在列):")
print(transposed_expression_data)# 3. 对转置后的矩阵执行PCA (这里只演示输入,不深入PCA细节)
# prcomp() 是R中进行PCA的函数
pca_results <- prcomp(transposed_expression_data, scale. = TRUE) # scale. = TRUE 标准化数据
print("PCA结果(部分):")
print(pca_results$x) # 显示主成分坐标# 思考:为什么PCA需要将矩阵转置?如果矩阵中存在字符型数据(比如不小心把某个表达量写成了"low"),prcomp函数能运行吗?

数据框(Data Frame):最常用的表格型数据结构
定义: 数据框是R中最常用、最灵活的表格型数据结构。它类似于一个电子表格,每列可以是不同的数据类型,但每列内的元素必须是同种类型。每行代表一个观察值,每列代表一个变量。
- 应用场景:
差异表达分析结果表: 一列是基因名(字符型),一列是log2FC(数值型),一列是P值(数值型),一列是FDR(数值型),一列是基因ID(字符型)等。
样本信息表: 样本ID、分组、年龄、性别等。各种统计分析的输入数据。 - 操作与理解:
使用data.frame()创建。
通过$符号或[]索引来访问列。
强大的数据筛选、排序、合并能力。
【实战演练:绘制火山图的数据准备与筛选】
火山图是差异表达分析中最常见的可视化图表,它结合了基因表达变化的倍数(Log2FC)和统计显著性(P值),帮助我们快速识别差异表达基因。火山图的绘制,正是数据框操作的典型应用。
场景: 我们得到了一份包含基因ID、log2FC和FDR值的差异表达分析结果。
# 1. 创建一个数据框,模拟差异表达分析结果
diff_expression_df <- data.frame(GeneID = c("GeneA", "GeneB", "GeneC", "GeneD", "GeneE", "GeneF", "GeneG", "GeneH"),log2FC = c(2.5, -1.8, 0.1, 3.0, -0.5, -2.2, 0.8, 1.5),FDR = c(0.001, 0.008, 0.85, 0.0001, 0.60, 0.002, 0.45, 0.015)
)
print("原始差异表达结果数据框:")
print(diff_expression_df)# 2. 添加一列 -log10(FDR),用于火山图Y轴,数值越大表示越显著
diff_expression_df$neglog10FDR <- -log10(diff_expression_df$FDR)
print("添加-log10(FDR)列后的数据框:")
print(diff_expression_df)# 3. 根据阈值筛选差异表达基因
# 筛选标准:log2FC绝对值大于2 且 FDR小于0.05
significant_genes <- subset(diff_expression_df, abs(log2FC) > 2 & FDR < 0.05)
print("筛选出的显著差异表达基因:")
print(significant_genes)# 4. 准备绘制火山图 (这里不实际绘制,只展示数据输入)
# 绘制火山图的函数(例如ggplot2::ggplot)会直接使用这个数据框的列
# library(ggplot2)
# ggplot(diff_expression_df, aes(x = log2FC, y = neglog10FDR)) +
# geom_point(aes(color = ifelse(abs(log2FC) > 2 & FDR < 0.05, "Significant", "Non-significant"))) +
# geom_vline(xintercept = c(-2, 2), linetype = "dashed", color = "grey") +
# geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "grey") +
# theme_minimal()
# 思考:如果GeneID列是数值型,或者log2FC列是字符型,会发生什么?为什么?

列表(List):最灵活的数据“容器”
定义: 列表是R中最灵活的数据结构,可以存储任意类型、任意长度的R对象。它可以是向量、矩阵、数据框,甚至可以是另一个列表。
- 应用场景:
单细胞分析对象: 单细胞分析中,一个Seurat对象或SingleCellExperiment对象,本质上就是一个大型列表,其中包含:
a. 一个稀疏矩阵(表达量数据)。
b. 一个数据框(细胞的元数据,如细胞类型、批次信息)。
c. 另一个数据框(基因的元数据)。
d. 多个矩阵(降维结果,如PCA、UMAP坐标)。
e. 多个列表(聚类结果、差异基因列表等)。
存储复杂的统计模型结果。一次性返回多个不同类型的结果。
- 操作与理解:
使用list()函数创建。
通过[[索引]]或$符号访问列表中的元素。
【实战演练:单细胞数据对象的抽象理解】
在单细胞RNA测序(scRNA-seq)分析中,我们通常不会直接操作原始矩阵,而是使用专门的R包(如Seurat或Bioconductor的SingleCellExperiment)构建一个复杂的“单细胞对象”。这个对象本质上就是一个包含多种数据结构的列表。
场景: 抽象模拟一个简化的单细胞数据对象,其中包含表达矩阵、细胞分组信息和降维坐标。
# 1. 模拟单细胞表达矩阵 (通常是稀疏矩阵,这里用普通矩阵简化)
# 5个基因,10个细胞
sc_expression_matrix <- matrix(sample(0:10, 50, replace = TRUE), # 随机生成0-10的整数表达量nrow = 5, ncol = 10,dimnames = list(paste0("Gene", 1:5),paste0("Cell", 1:10))
)# 2. 模拟细胞的元数据(例如细胞类型)
cell_metadata <- data.frame(CellID = paste0("Cell", 1:10),CellType = factor(c(rep("T_cell", 4), rep("B_cell", 3), rep("Macrophage", 3))),Batch = factor(c(rep("Batch1", 5), rep("Batch2", 5)))
)# 3. 模拟降维后的UMAP坐标
umap_coordinates <- matrix(rnorm(20), # 2列,10行,代表10个细胞的2D UMAP坐标nrow = 10, ncol = 2,dimnames = list(paste0("Cell", 1:10), c("UMAP_1", "UMAP_2"))
)# 4. 将这些数据组织成一个列表,模拟单细胞对象
single_cell_object <- list(counts = sc_expression_matrix,metadata = cell_metadata,reductions = list(umap = umap_coordinates),analysis_parameters = "Seurat_v5"# 可以存储分析版本等额外信息
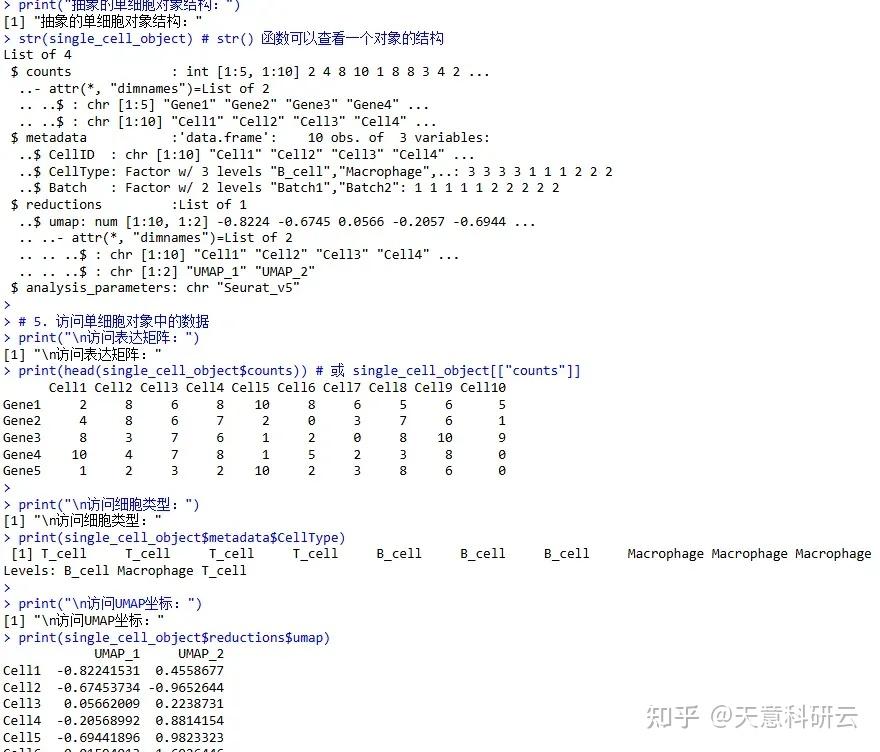
)print("抽象的单细胞对象结构:")
str(single_cell_object) # str() 函数可以查看一个对象的结构# 5. 访问单细胞对象中的数据
print("\n访问表达矩阵:")
print(head(single_cell_object$counts)) # 或 single_cell_object[["counts"]]print("\n访问细胞类型:")
print(single_cell_object$metadata$CellType)print("\n访问UMAP坐标:")
print(single_cell_object$reductions$umap)# 思考:为什么单细胞对象要设计成列表这种结构,而不是直接用一个大矩阵或数据框?
# 提示:考虑到不同类型的数据(数值、字符、分类),以及不同维度的数据(基因数x细胞数,细胞数x特征数)。
总结与展望:有序数据,高效分析
通过今天的学习和实践,你现在应该对R语言的数据类型和数据结构有了更深刻的认识。我们通过转录组的差异表达分析和火山图绘制,以及单细胞数据处理和PCA的数据准备,看到了向量、矩阵、数据框和列表如何在实际生信项目中发挥核心作用。
- 数据类型是数据的属性,它决定了数据能进行哪些操作。
- 数据结构是数据的组织形式,它决定了数据如何被高效存储和访问。
- R Project则为这些有序的数据提供了一个规范的“家”,保证了分析的可重复性。
思考题答案提示:
- 向量P值校正问题: 如果
raw_p_values中混入了字符型数据,p.adjust函数会报错,因为该函数期望所有输入都是数值型。R的向量是同质的,如果强制将数值和字符混合,所有元素会被强制转换为字符型。 - PCA矩阵转置问题:
prcomp()函数默认期望输入数据的行是观测值(通常是样本),列是变量(通常是基因)。因此,如果你的原始矩阵是基因在行、样本在列,就需要转置。如果矩阵中存在字符型数据,prcomp()函数会报错,因为它只能处理数值型数据。 - 火山图数据框问题: 如果
GeneID列是数值型,虽然可能不会立刻报错,但在后续如果需要进行基因名匹配等操作时会出错。如果log2FC列是字符型,abs()函数和subset()函数的数值比较会报错,因为它们期望数值型输入。 - 单细胞对象列表设计问题: 单细胞对象之所以设计成列表,是因为它需要在一个单一的结构中存储多种不同类型和不同维度的数据。表达矩阵是数值型二维数据,细胞元数据是包含多种类型的二维数据框,降维坐标是数值型二维数据,而分析参数可能是字符型或数值型。列表的异质性和嵌套性,使其成为容纳这些复杂且多样化数据的理想“容器”。如果使用单一矩阵或数据框,则无法同时满足所有数据的存储需求。