【论文阅读】ACE: Explaining cluster from an adversarial perspective

论文地址:ACE: Explaining cluster from an adversarial perspective (mlr.press)
摘要
单细胞RNA测序分析中一个常见的工作流程是:将数据投影到一个潜在空间中,在该空间中对细胞进行聚类,并识别一组标记基因,用以解释所发现聚类之间的差异。
这种三步流程的主要缺点在于各步骤彼此独立执行,从而忽略了非线性嵌入以及基因间依赖性对标记基因选择的影响。
在此,我们提出了一个集成的深度学习框架,称为对抗聚类解释(Adversarial Clustering Explanation, ACE),
它将这三个步骤整合为一个统一的流程。
该方法因此跳出了传统“标记基因”的概念,转而识别一组解释性基因面板。
这组基因面板可能包括:
不仅在某些细胞类型中富集,还在其他细胞类型中缺失的基因,
以及在高度相似的细胞类型之间表现出差异的基因。
在实证上,我们展示了ACE能够识别出既具有高度区分性又非冗余的基因面板,
并进一步展示了ACE在图像识别任务中的适用性。
引言
以下是你提供的整段英文内容的完整中文翻译:
单细胞测序技术使得我们能够以高通量方式研究基因组生物学的多个方面,包括基因表达、DNA甲基化、组蛋白修饰、染色质可及性和基因组三维结构(Stuart & Satija, 2019)。在这些应用中,所得的高维数据通常可以表示为一个稀疏矩阵,其中行对应于单个细胞,列对应于这些细胞的特征(如基因表达值、甲基化事件等)。实证研究表明,这些数据分布在一个具有潜在语义结构的低维流形上(Welch et al., 2017)。因此,以潜在语义为基础识别细胞群体,并分析这些群体之间的差异,是该领域的一个重要研究方向(Plumb et al., 2020)。
在本研究中,我们聚焦于**单细胞RNA测序(scRNA-seq)**数据的分析。scRNA-seq 是最广泛可获得的单细胞测序数据类型,其分析具有挑战性,不仅因为数据的高维性,还因为其中存在噪声、批次效应和稀疏性(Amodio et al., 2019)。scRNA-seq 数据本身表现为一个稀疏的“细胞×基因”矩阵,通常包含数万到几十万个细胞,以及数万个基因。
scRNA-seq 分析中常见的工作流程包括以下三个步骤(Pliner et al., 2019):
-
将细胞投影到低维空间中,学习其紧凑表示;
-
在该低维表示中识别相似细胞群体(通常通过聚类实现);
-
比较各个群体之间的基因表达差异,以理解每个群体对应的生物学过程。
此外,还可以使用已知的“标记基因”将这些细胞群体标注为具体细胞类型。
这个三步流程的主要缺点在于每个步骤都是独立执行的。为此,我们提出了一个集成的深度学习框架 ACE(Adversarial Clustering Explanation),用于scRNA-seq数据的统一分析流程。ACE 能够将数据投影到潜在空间,对细胞进行聚类,并识别出能简明解释不同聚类之间差异的一组基因(见图1)。
从高层次来看,ACE 首先将聚类过程“神经网络化”,即将其重构为一个功能等价的多层神经网络(Kauffmann et al., 2019)。结合用于生成低维表示的深度自编码器,ACE 能够利用基于梯度的神经网络解释方法,将细胞的聚类归属追溯到输入基因。
接下来,ACE 针对每个样本,寻找其输入基因表达谱的小扰动,这些扰动可以促使神经网络模型改变对该样本的聚类归属。这些对抗扰动使 ACE 能够为每个聚类(或一对聚类)定义一组精炼的基因特征集。
ACE 试图回答如下问题:
“对于某个特定细胞群体,是否可以识别出一小组基因,其表达模式足以区分该群体的成员?”
我们将该问题建模为一个排序任务,通过对排序结果设定阈值,从而得到解释性基因集合。
ACE 的联合建模方法相比于现有技术具有多方面优势:
-
现有大多数方法在分析流程的第三步——识别群体相关基因——时,通常是逐个基因独立分析(Love et al., 2014),忽略了由基因调控网络诱导的基因间依赖关系,导致所识别的基因集合高度冗余。相比之下,ACE 旨在找出能联合解释某个聚类(或聚类对)的一小组基因。
-
多数方法在识别群体相关基因时,并不考虑非线性嵌入模型对数据的映射作用——也就是忽略了正是该嵌入模型定义了聚类空间。而目前唯一的例外是 GCE(Global Counterfactual Explanation) 算法(Plumb et al., 2020),但它仅支持线性变换。
-
ACE 的集成方法还能在基因指派过程中考虑批次效应。像 t-SNE(Van der Maaten & Hinton, 2008)和 UMAP(McInnes & Healy, 2018; Becht et al., 2019)等标准非线性嵌入方法不能处理此类结构信息,可能导致对数据的错误解释(Amodio et al., 2019; Li et al., 2020)。为应对这一问题,可使用集成去噪与批次校正的深度自编码器进行分析(Lopez et al., 2018; Amodio et al., 2019; Li et al., 2020)。我们在下文中展示了如何将批次效应结构有效集成进 ACE 模型。
ACE 的一个显著特征是:通过联合识别基因,该方法不再依赖传统“标记基因”的概念,而是提出了“基因面板(gene panel)”的概念。
因此,面板中的基因可能并不全都在某个聚类中富集,而是它们共同对该聚类具有预测性。
更具体地说,ACE 不仅给出基因的排序结果,还为每个基因分配一个布尔值,用于指示其在该面板中的作用是正向(富集)还是负向(缺失),即该基因的表达是否相对于群体归属被增强或被削弱。
我们在模拟数据与真实数据集上验证了 ACE 的有效性,实验证明 ACE 能够识别出具有高度区分性和低冗余性的基因面板。此外,我们还展示了 ACE 在非生物领域(如图像识别)中的应用潜力。
方法
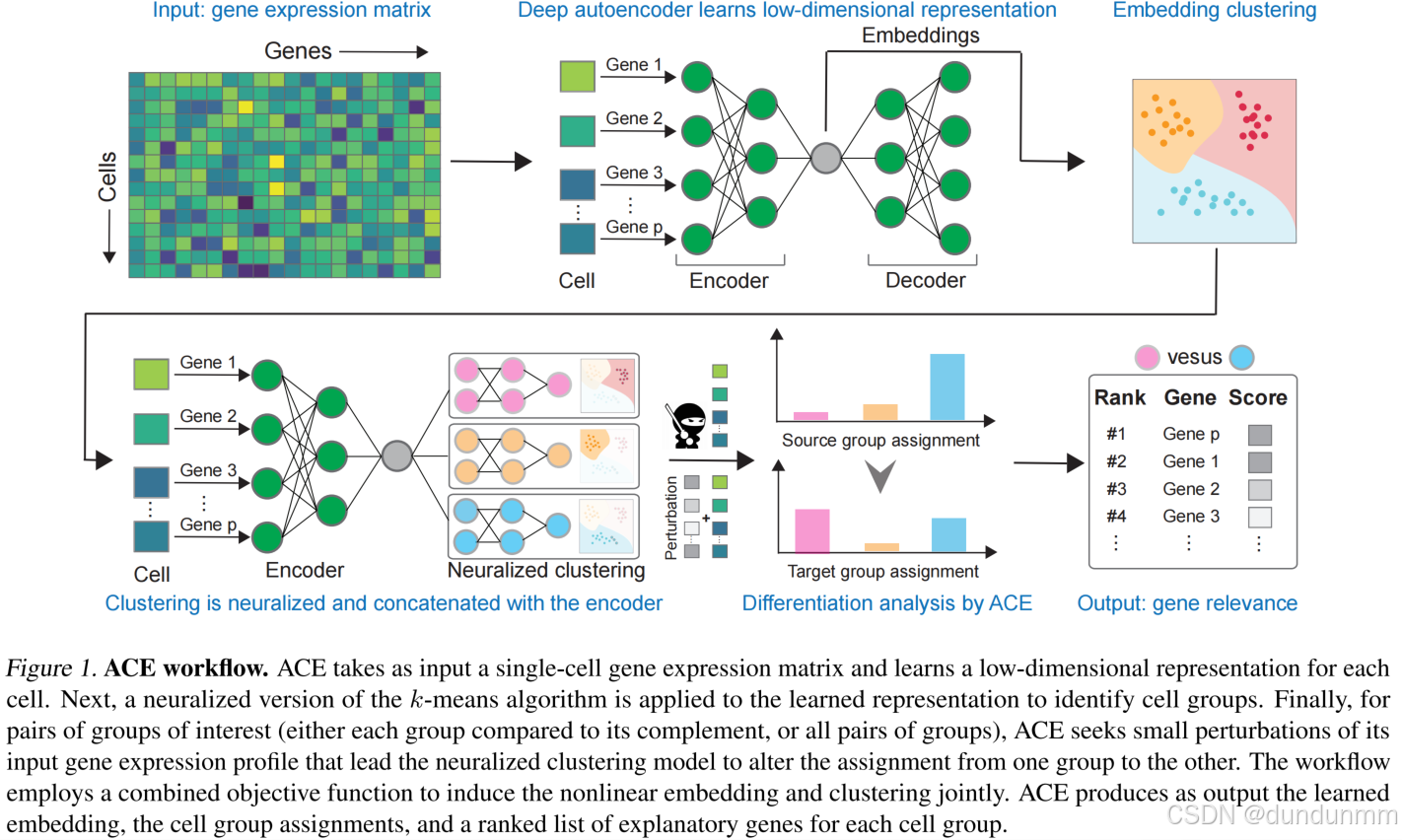
前面就是常规的表示学习,主要关注群体解释(Explaining the groups)”部分的技术细节:
群体解释(Explaining the groups)
ACE 的最后一步旨在为神经网络化的 k-means 聚类算法所识别出的每个聚类诱导一个基因排序,其中得分较高的基因最能解释该聚类。
我们考虑该任务的两种变体:
-
一对其余(one-vs-rest)设置:将关注的群体 Zs=f(Xs)⊆Z与其补集 Zt=f(Xt)⊆Z进行比较,其中 Xt=X∖Xs;
-
一对一(one-vs-one)设置:将一个关注群体 Zs=f(Xs)⊆Z与另一个关注群体 Zt=f(Xt)⊆Z进行比较。
在这两种设置中,目标是识别出输入空间(即基因表达空间)中源群体 Xs⊆X 与目标群体 Xt⊆X之间的关键差异。
我们将该问题视为一个神经网络解释任务,其核心思想是在群体内寻找最小扰动,使得样本 x∈Xsx的聚类归属从源群体 s改变为目标群体 t。
具体来说,我们优化的目标函数由两个部分组成:
-
当前样本 x与扰动后样本 x^=x+δ之间的差异(鼓励扰动尽可能小);
-
扰动导致的聚类归属变化的显著性(鼓励扰动后的样本被正确地划分为目标群体)。
🎯 一对一设置的目标函数如下:
![]()
其中:
-
∥δ∥1是对扰动大小的L1正则,用于鼓励扰动稀疏、特征非冗余;
-
λ>0是权衡因子,当其取值较小时鼓励小幅扰动,当其较大时则更关注目标群体归属的实现;
-
第二项惩罚源群体的分类得分(logit)在扰动后仍大于目标群体的情形;
-
α>0是预设的 margin(本文中设为 α=1.0);
-
假设输入表达矩阵已归一化,使得不同基因上的扰动具有可比性。
🎯 一对其余设置的目标函数如下:
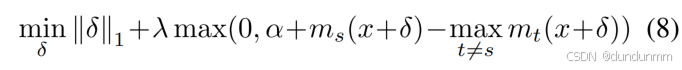
其中第二项惩罚的是:源群体的logit值依然大于所有非源群体的最大logit值的情况。
🧬 基因重要性排序:
通过优化上面任一目标函数(公式7或8)获得的扰动向量 δ∈Rp,
ACE 将第 i 个基因的重要性定义为扰动值的绝对值 ∣δi∣,
从而建立一个基因排序,值越大的基因被认为对该群体越具有特异性。
实验
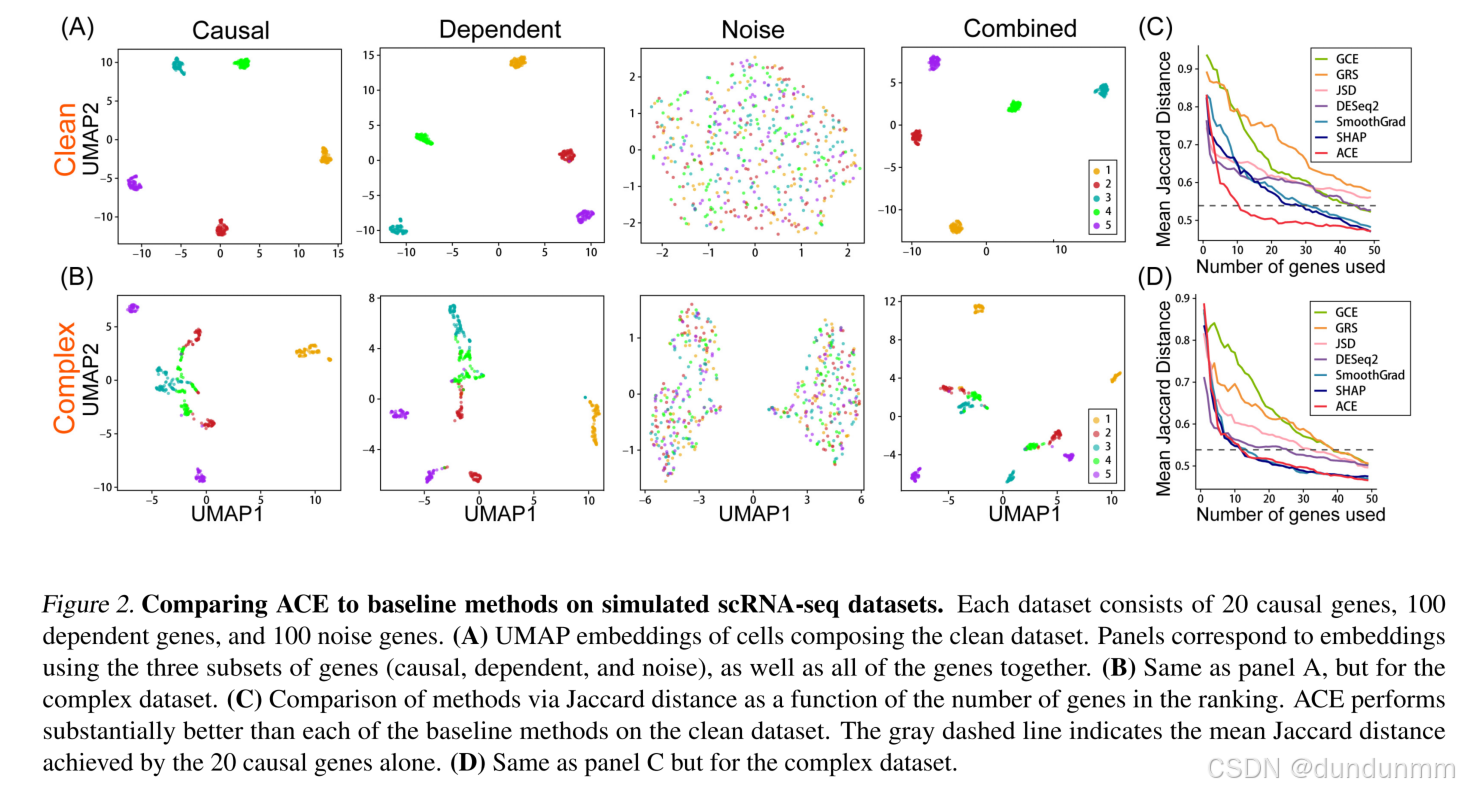
这篇发表的较早。