作者,Evil Genius
本篇是一些总结,为基因组的内容做一些准备。
单细胞空间检测CNV、和基因组WGS/WES检测CNV都是分析中常见的内容,其中单细胞空间常用的方法是inferCNV、inferCNVpy、SpatialinferCNV等,而WGS/WES常用的是cnvkit、GATK、VarScan2。
我们来汇总一下关于CNV的检测方法, 最后比较一下单细胞空间与基因组检测CNV的差异。
总结完就知道为什么现在都要求上多组学了,而且单细胞 + WES是目前最经济的多组学方案。
先来看单细胞空间的部分
1. inferCNV(经典单细胞CNV推断工具)
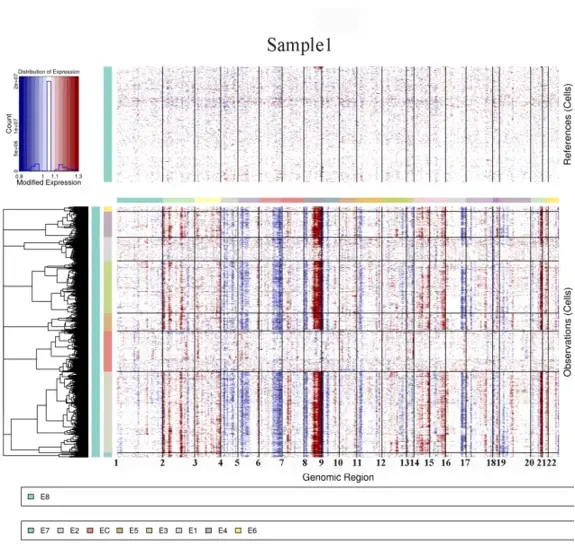
方法原理
inferCNV 是最早用于 scRNA-seq 数据 CNV 检测的 R 包,其核心思路是:
- 参考表达谱构建:使用正常细胞(如免疫细胞、基质细胞)的基因表达作为基线参考。
- 基因组滑动窗口平滑:按染色体位置排序基因,计算每个细胞在滑动窗口(默认100个基因)内的平均表达量。
- CNV信号提取:比较肿瘤细胞与参考细胞的表达差异,推断拷贝数变化(缺失/正常/扩增)。
- 聚类分析:使用层次聚类或Leiden算法识别具有相似CNV模式的细胞亚群。
- **HMM(隐马尔可夫模型):预测离散的CNV状态(如单拷贝缺失、双拷贝缺失、扩增等)。
课程上我们讲解的更加详细

包括阈值的选择

关键参数
(1) 数据预处理
| 参数 | 功能 | 默认值 |
|---|
| cutoff | 基因表达量阈值(过滤低表达基因) | 0.1 |
| min_cells_per_gene | 基因至少在多少细胞中表达 | 3 |
| num_ref_groups | 参考细胞分组数(提高参考稳健性) | 3 |
(2) CNV计算
| 参数 | 功能 | 默认值 |
|---|
| window_length | 滑动窗口大小(基因数) | 100 |
| smooth_method | 平滑方法("pyramidinal"或"runmeans") | "pyramidinal" |
| max_centered_threshold | 中心化截断值(控制极端值) | 3 |
(3) 聚类方法
| 参数 | 功能 | 选项 |
|---|
| cluster_method | 聚类算法 | "hclust"(层次聚类)或 "leiden"(社区检测) |
| leiden_resolution | Leiden聚类的分辨率(值越大,亚群越多) | 1.0 |
| hclust_method | 层次聚类方法("ward.D", "complete"等) | "ward.D" |
(4) HMM(可选)
| 参数 | 功能 | 默认值 |
|---|
| HMM | 是否运行HMM预测CNV状态 | FALSE |
| HMM_type | HMM类型("i3"=3状态,"i6"=6状态) | "i3" |
| tumor_subcluster_pval | 肿瘤亚群显著性阈值 | 0.1 |
2. inferCNVpy(Python优化版)
方法原理
inferCNVpy 是 inferCNV 的 Python 实现,优化了计算效率,并增加了 UMAP/t-SNE 可视化 功能。
- 支持 AnnData 对象,与 Scanpy 生态无缝集成。
- 提供 交互式可视化(如 Plotly 动态热图)。
- 计算更快,适合 大规模单细胞数据集。
参加过培训班的应该知道,这个方法目前已经被多篇高分文献采用。

关键参数
(1) 数据预处理
| 参数 | 功能 | 默认值 |
|---|
| min_genes | 每个细胞至少检测到的基因数 | 200 |
| min_cells | 每个基因至少表达的细胞数 | 3 |
| reference | 参考细胞类型(如免疫细胞) | None(需用户指定) |
(2) CNV计算
| 参数 | 功能 | 默认值 |
|---|
| window_size | 滑动窗口大小 | 100 |
| smooth_rolling | 是否使用滚动平均平滑 | True |
| max_val | 表达量截断值(防止极端值影响) | 3 |
(3) 降维与可视化
| 参数 | 功能 | 选项 |
|---|
| use_umap | 是否对CNV矩阵进行UMAP降维 | True |
| use_tsne | 是否进行t-SNE降维 | False |
| plot_interactive | 是否生成交互式热图(Plotly) | True |
SpatialInferCNV(空间转录组CNV检测)
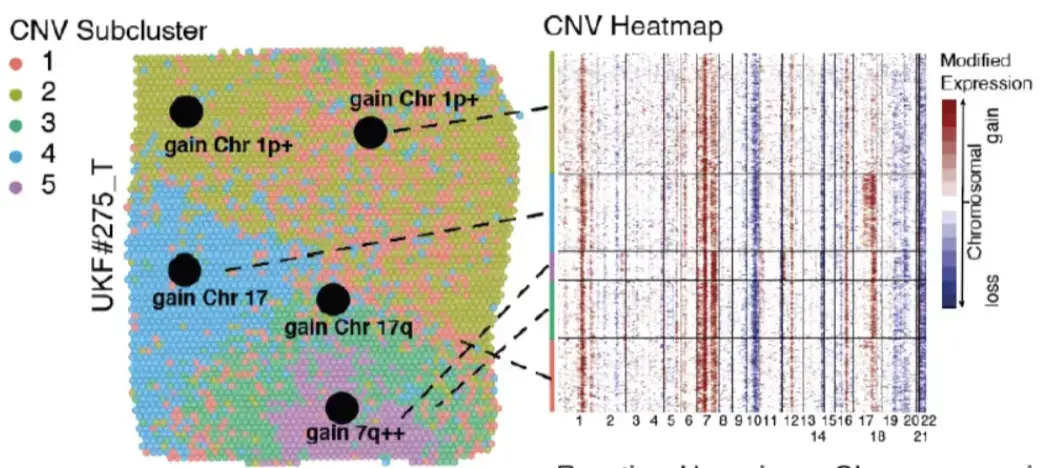
方法原理
SpatialInferCNV 在 inferCNV 基础上 整合空间信息,适用于 Visium等空间转录组数据:
- 空间约束平滑:在基因组滑动窗口基础上,加入 空间邻域平滑(如10× Visium的spot邻近关系)。
- 肿瘤边界识别:利用CNV模式划分 肿瘤区域 vs. 正常组织。
- 空间热点检测:识别基因组变异的空间聚集模式(如局部扩增)。
关键参数
| 参数 | 功能 | 默认值 |
|---|
| spatial_weight | 空间平滑权重(0=仅基因组,1=强空间约束) | 0.5 |
| neighborhood_size | 空间邻域范围(spot数量) | 6 |
| detect_boundaries | 是否计算肿瘤边界 | True |
| hotspot_pval | 空间CNV热点的显著性阈值 | 0.01 |
4. 方法对比与总结
| 方法 | 适用数据 | 核心算法 | 关键功能 | 计算速度 |
|---|
| inferCNV | scRNA-seq | 滑动窗口 + HMM | 层次/Leiden聚类 | 中等 |
| inferCNVpy | scRNA-seq | 优化滑动窗口 | UMAP/交互式热图 | 快 |
| SpatialInferCNV | 空间转录组 | 基因组+空间平滑 | 肿瘤边界、热点检测 | 中等 |
适用场景
- inferCNV:经典方法,适合小规模scRNA-seq数据,需R环境。
- inferCNVpy:Python用户首选,适合大数据集,支持UMAP可视化。
- SpatialInferCNV:空间转录组数据,研究肿瘤微环境。
总结
- 无参考数据? 可尝试 CopyKAT(基于全局表达模式)。
- 需要空间信息? 选择 SpatialInferCNV。
- 大规模数据? 推荐 inferCNVpy(计算更快)。
再来看WES/WGS的部分
1. CNVkit(基于测序深度和靶向测序优化)

方法原理
CNVkit 是一种灵活的工具,适用于 WGS 和 WES 数据,尤其擅长 靶向测序(如panel测序) 的CNV检测。其核心步骤包括:
- 靶向区域覆盖度计算:统计每个目标区域的测序深度。
- 参考样本归一化:使用对照样本(如正常组织或公共数据库)校正技术偏差(GC含量、比对效率等)。
- 分段(segmentation):使用 CBS(Circular Binary Segmentation) 或 HMM(Hidden Markov Model) 算法划分CNV区域。
- 调用CNV:基于log2比值和统计显著性判断拷贝数变化(缺失/扩增)。
关键参数
| 参数 | 功能 | 默认值/选项 |
|---|
| --method | 分段算法 | cbs(默认)hmm、none |
| --drop-low-coverage | 过滤低覆盖区域 | False(建议WES设为True) |
| --normal | 对照样本(用于归一化) | 需用户提供 |
| --targets | 目标区域(BED文件) | 必须提供(WES/panel测序) |
| --antitargets | 非目标区域(用于WGS) | 可选 |
| --min-variant-depth | 最低支持深度 | 20 |
适用场景:
- WES/靶向测序(如肿瘤panel测序)
- 需要高灵敏度的小CNV检测
- 无匹配正常样本时,可使用公共数据库参考
2. GATK(GermlineCNVCaller & ModelSegments)
方法原理
GATK(Genome Analysis Toolkit)提供了 GermlineCNVCaller(种系CNV)和 ModelSegments(体细胞CNV)两种方法:
- GermlineCNVCaller(适用于种系CNV):
- 基于 Poisson Latent Factor Model(PLFM) 校正批次效应和GC偏差。
- 使用 Gaussian Mixture Model(GMM) 分段。
- 输出 CNV片段和置信度评分。
ModelSegments(适用于肿瘤-正常配对样本):
- 计算肿瘤/正常样本的 log2比值。
- 使用 HMM 预测CNV状态(如单拷贝缺失、扩增等)。
- 可结合 Allelic Fraction(等位基因频率) 提高准确性。
关键参数
| 参数 | 功能 | 默认值/选项 |
|---|
| --model | 模型选择 | POISSON(GermlineCNVCaller)、HMM(ModelSegments) |
| --number-of-samples | 样本数(用于批次校正) | 需用户指定 |
| --minimum-total-allele-count | 最低等位基因计数 | 10 |
| --call-threshold | CNV调用阈值 | 0.5(log2 ratio) |
| --kernel-variance | HMM核方差(影响灵敏度) | 0.25 |
适用场景:
- 种系CNV检测(GermlineCNVCaller)
- 肿瘤-正常配对样本(ModelSegments)
- 需要高精度CNV边界检测
3. VarScan2(基于测序深度和统计检验)
方法原理
VarScan2 最初设计用于 SNP/Indel检测,但其 copyCaller 模块可用于CNV分析:
- 计算覆盖深度:统计目标区域的测序深度。
- 归一化:校正GC偏差和比对效率。
- 统计检验:使用 Kolmogorov-Smirnov(KS)检验 判断CNV是否显著。
- CNV调用:基于 log2比值 和 p值 判断缺失/扩增。
关键参数
| 参数 | 功能 | 默认值/选项 |
|---|
| --min-coverage | 最低覆盖深度 | 20 |
| --min-mean-rd | 最低平均测序深度 | 10 |
| --p-value | KS检验显著性阈值 | 0.01 |
| --data-ratio | 肿瘤/正常深度比值阈值 | 0.8(缺失)、1.2(扩增) |
| --somatic-p-value | 体细胞CNV的p值阈值 | 0.05 |
适用场景:
- 肿瘤-正常配对样本(体细胞CNV检测)
- 快速分析(计算量较小)
- 需要统计显著性评估
方法对比与汇总
| 工具 | 核心算法 | 适用场景 | 优点 | 局限性 |
|---|
| CNVkit | CBS/HMM + 归一化 | WES/靶向测序 | 高灵敏度,支持无对照样本 | 依赖目标区域(WGS需额外参数) |
| GATK | PLFM/HMM + 等位基因频率 | 种系/体细胞CNV | 高精度,整合GATK生态 | 计算量大,依赖正常对照 |
| VarScan2 | KS检验 + log2比值 | 肿瘤-正常配对 | 计算快,统计检验严格 | 仅适用于配对样本 |
总结
- WES/靶向测序 → CNVkit(灵活,灵敏度高)
- 种系CNV → GATK GermlineCNVCaller(批次校正能力强)
- 肿瘤-正常配对 → GATK ModelSegments 或 VarScan(VarScan更轻量)
- 需要统计显著性 → VarScan2(KS检验提供p值)
最后来总结一下单细胞空间和基因组检测CNV的核心差异
数据来源与分辨率
| 维度 | 单细胞/空间转录组 | 基因组(WGS/WES) |
|---|
| 数据基础 | 基于RNA表达量(间接推测CNV) | 基于DNA测序深度/结构变异(直接检测CNV) |
| 分辨率 | 单细胞级(~10μm空间分辨率) | 群体水平(混合细胞) |
| 覆盖范围 | 仅检测表达基因(受转录活性限制) | 全基因组/外显子组覆盖 |
检测原理差异
| 方法 | 单细胞/空间转录组 | 基因组(WGS/WES) |
|---|
| 核心信号 | 基因表达异常(如高表达基因簇提示扩增) | DNA测序深度异常/断点检测 |
| 参考基准 | 依赖正常细胞表达谱(如免疫细胞) | 依赖对照样本或公共数据库(如1000 Genomes) |
| 算法重点 | 滑动窗口平滑 + 相对表达差异 | 统计模型(如HMM/CBS) + 读段分析(PE/SR) |
技术挑战与局限
| 挑战 | 单细胞/空间转录组 | 基因组(WGS/WES) |
|---|
| 假阳性来源 | 转录噪声(如基因爆发表达) | 测序偏差(如GC含量/比对效率) |
| 灵敏度限制 | 仅能检测转录活跃区域的CNV | 可检测非编码区/沉默区域CNV |
| 空间特异性 | 可解析肿瘤微环境的空间异质性(仅空间转录组) | 无法提供空间信息 |
应用场景对比
| 场景 | 单细胞/空间转录组 | 基因组(WGS/WES) |
|---|
| 肿瘤异质性 | 识别亚克隆结构 + 空间分布(如侵袭前沿) | 检测克隆性CNV驱动事件 |
| 遗传病研究 | 不适用 | 种系CNV检测(如微缺失综合征) |
| 技术互补性 | 需结合基因组数据验证功能性CNV | 提供金标准,但缺乏单细胞分辨率 |
核心结论
直接性 vs. 间接性
- WGS/WES直接检测DNA层面的CNV,结果更可靠;
- 单细胞方法通过RNA表达间接推断,受转录调控干扰。
分辨率权衡
- 单细胞/空间数据提供细胞级分辨率但覆盖有限;
- WGS/WES提供全基因组覆盖但无法区分单个细胞。
最佳实践建议
- 先导分析:用WGS/WES鉴定高置信度CNV;
- 精细解析:用单细胞/空间数据研究异质性和功能影响;
- 多组学整合:联合DNA+RNA数据提高CNV生物学解释力(如扩增基因是否真实高表达)。
生活很好,有你更好