链特异性文库是什么?为什么它在转录组测序中越来越重要?
链特异性文库是什么?为什么它在转录组测序中越来越重要?
在现代分子生物学研究中,RNA测序(RNA-seq) 是一种广泛应用的技术,用于分析基因在不同条件下的表达情况。而在RNA-seq的众多技术细节中,有一个“隐秘但关键”的环节——链特异性文库构建(Strand-specific library preparation)。这项技术虽然听起来有些专业,但它对结果的准确性有着重要影响。本文将通俗地介绍链特异性文库的原理、作用、常见方法及数据分析注意事项。
1. 什么是“链特异性”?
DNA是一种双链螺旋结构,由一条正义链(+链)和一条反义链(–链)构成。转录过程中,通常是由DNA的反义链(–链)作为模板合成mRNA,从而使mRNA序列与正义链一致(除了碱基T被替换为U)。
而在传统的RNA-seq文库构建中,RNA被打断后逆转录成cDNA,再建库测序,这个过程不会记录RNA是来源于哪一条DNA链的信息。我们只知道这段RNA存在,但不知道其是源于正链还是反链。
链特异性文库构建的目标是,在建库过程中通过特定方法保留RNA原始的转录方向性信息,从而区分每一条RNA是由正链还是反链转录来的。
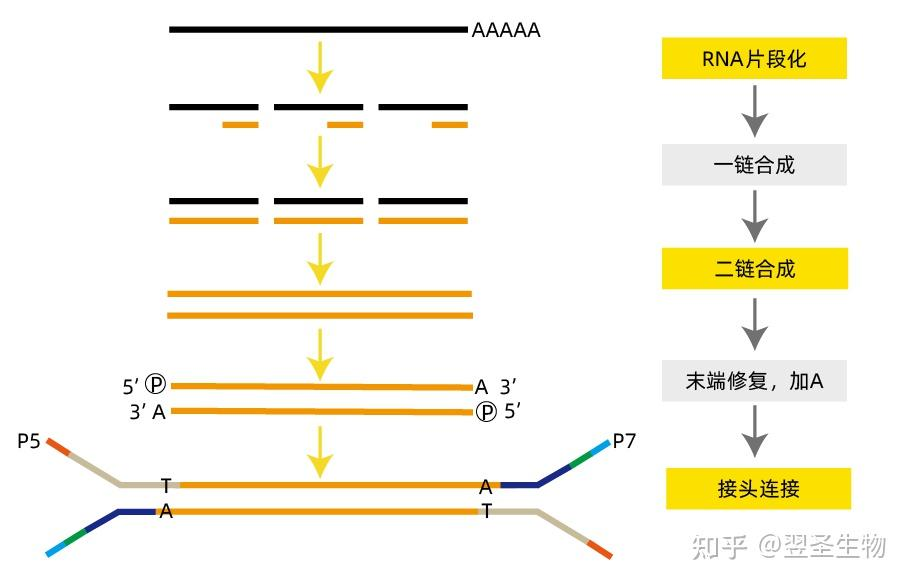
2. 为什么需要链特异性文库?
链方向的保留,在多种分析中具有不可替代的重要性:
区分重叠基因
部分基因在基因组中是反向重叠的,即它们位于同一个基因组区域的两条链上。如果没有链信息,无法准确判断这段表达信号来自哪个基因。
注释非编码RNA
例如lncRNA、反义转录本等非编码RNA,常与编码基因反向重叠。链信息是这些转录本精确注释的关键。
提高定量精度
当多个基因之间位置相近或有部分重叠时,链特异性测序可显著减少表达混淆,提高定量和差异分析的准确性。
3. 链特异性文库的实现原理
主流链特异性文库构建方法主要分为以下几类,它们的共同目标是在建库过程中保留或标记RNA的方向性信息。
方法一:dUTP法(Illumina常用方案)
dUTP法是目前最常用的链特异性建库策略,原理如下:
- 合成第一链cDNA(使用mRNA为模板)
- 合成第二链时,用dUTP代替dTTP,使第二链中含有尿嘧啶(dU)
- 使用**UDG(Uracil-DNA Glycosylase)**选择性降解含dU的第二链
- 仅保留第一链进行接头连接与PCR建库
此法操作简单、成本低、兼容性好,是Illumina TruSeq等商业试剂盒的推荐方案。
注意事项:
- 测序得到的read方向与原始mRNA方向相反
- 常用参数方向性为
RF(Read1为反义)
方法二:接头定向连接法(如 SMARTer、ScriptSeq)
通过在第一链cDNA末端引入方向性接头或模板切换寡核苷酸(TSO),实现链信息的标记。例如:
- SMARTer法:只在第一链延伸出接头,方向性由其控制。适用于低输入甚至单细胞RNA。
- ScriptSeq法:通过特定引物和接头组合区分方向,较早用于链特异性建库。
方法三:标签标记法(Ligation-based)
该法通过在cDNA两端连接不同标签序列来区分方向性,部分早期方案采用,但操作复杂,使用较少。
4. 如何判断文库是否为链特异性?
在测序实验前或数据分析时,应确认建库是否保留方向信息,可通过以下方法判断:
- 查看实验说明书或FastQC注释,如“stranded = yes”
- 使用RSeQC工具(infer_experiment.py) 判断read是否集中来源于特定链
- 检查比对软件中strand参数是否正确设置,避免方向误判
5. 链特异性数据的分析注意事项
分析链特异性RNA-seq数据时,需明确方向性设定:
| 分析步骤 | 重点参数 | 示例说明 |
|---|---|---|
| 比对软件 | 设置strand参数 | HISAT2示例:--rna-strandness RF |
| featureCounts计数工具 | 设定链信息 | -s 1为正链,-s 2为反链(dUTP法用-s 2) |
| HTSeq-count工具 | 设置为reverse方向 | -s reverse |
| 定量分析 | 匹配注释方向 | lncRNA尤其敏感,方向错会导致显著误判 |
6. 建库方案选择建议与参数配置
建库方法建议
| 研究目标 | 建议建库方案 | 说明 |
|---|---|---|
| mRNA表达分析 | dUTP法(TruSeq) | 成熟稳定、性价比高 |
| 非编码RNA分析(lncRNA等) | dUTP法或SMARTer法 | 保留方向,适合复杂转录本识别 |
| 单细胞或低起始量样本 | SMARTer、NEBNext Ultra II | 高灵敏度,适合微量RNA |
| 全转录组/非polyA分析 | rRNA去除 + dUTP法 | 可识别非polyA RNA转录本 |
实验参数配置参考(以dUTP法为例)
| 步骤 | 参数或建议 |
|---|---|
| RNA输入量 | 100 ng – 1 µg,依样品而定 |
| 打断条件 | 94°C,4–8分钟,目标片段200–400 bp |
| 第一链合成 | 使用SuperScript II或III等高效酶 |
| 第二链合成 | 用dUTP替代dTTP |
| 降解第二链 | 使用USER酶去除含dU链 |
| PCR扩增 | 控制在10–15个cycle内 |
| 文库质控 | Bioanalyzer检测片段分布峰值约300 bp |
7. 数据分析参数设置示例
HISAT2 比对示例
hisat2 -x genome_index -1 R1.fastq -2 R2.fastq --rna-strandness RF
其中 RF 表示链特异性双端测序,第一条read与mRNA方向相反。
STAR 比对配置
--outSAMstrandField intronMotif
--outSAMtype BAM SortedByCoordinate
--outFilterMultimapNmax 1
--twopassMode Basic
STAR支持链方向性,但后续需在featureCounts中设定方向。
featureCounts 示例
featureCounts -s 2 -p -T 8 -a annotation.gtf -o counts.txt aligned.bam
-s 2 表示反向链特异性,适用于dUTP建库。
HTSeq-count 示例
htseq-count -f bam -s reverse -r pos aligned.bam annotation.gtf
8. 如何验证链特异性是否有效
使用RSeQC工具包中的 infer_experiment.py 命令可以判断测序数据是否保留链信息:
infer_experiment.py -i aligned.bam -r ref.bed
结果会输出reads在不同链的分布比例。若某一类链向占比超过95%,说明链特异性建库成功:
Fraction of reads explained by "1++,1--,2+-,2-+": 0.958
Fraction of reads explained by "1+-,1-+,2++,2--": 0.042
9. 常见方法与参数汇总
| 方法类型 | 建库原理 | 分析参数方向性 | 适用场景 |
|---|---|---|---|
| dUTP法 | 第二链含dUTP并降解 | RF 或 -s 2 | 主流方案,Illumina推荐 |
| SMARTer法 | 模板切换接头控制方向性 | 需自定义 | 低起始量或单细胞样本 |
| Ligation-based法 | 接头序列标记方向性 | 需自定义 | 特殊需求项目,较复杂较少使用 |
小结
链特异性文库技术为RNA-seq分析带来了更高的准确性,特别适用于区分反向重叠基因、识别非编码RNA及提高定量精度。尽管建库成本略有增加、分析参数需设定更精确,但其带来的数据质量提升远大于投入。如果你正计划开展转录组研究,链特异性文库无疑是值得优先选择的建库方式之一。
如需配套文库构建图示、参数设定流程图、分析代码封装,欢迎留言交流。