【论文阅读】Nonparametric clustering of RNA-sequencing data

论文地址:Nonparametric clustering of RNA‐sequencing data (wiley.com)
代码地址:GitHub - Matematikoi/non_parametric_clustering: A repository containing nonparametric clustering algorithms from the manuscript "Nonparametric clustering for RNA-seq data" by Michael Levine, Nadia Atallah Lanman, Gabriel Lozano
摘要
在转录组数据中识别共表达基因的聚类是一项具有挑战性的任务。用于该目的的大多数算法通常可以归为两大类:基于距离的方法或基于模型的方法。
基于距离的方法通常利用数据对象之间的距离函数,并将相似的对象聚合为同一簇。而基于模型的方法则建立在混合模型框架的基础上。相比于基于距离的方法,基于模型的方法在解释性方面更具优势,因为每个聚类都可以通过所建立的模型明确刻画。
然而,这类方法的一个主要难点在于:很难确定一个适合用于混合模型的多元分布形式。
在本文中,我们首先回顾了用于选择混合模型中分布的一些现有方法。随后,我们提出采用非参数的最大平滑似然(MSL)算法来完全绕过这一问题。该算法此前已在统计学文献中被提出,但据我们所知,尚未在转录组数据中得到应用。
这种方法的一大特点在于:无需明确指定每个生物样本的分布形式,从而简化了实际操作过程。
我们在模拟数据上进行了实验,并将该算法应用于两个真实数据集。结果显示:当应用于真实数据时,该算法可以识别出大量具有生物学意义的聚类,并且其表现至少不逊于当前常用于RNA-seq数据聚类的其他几种混合模型算法。
我们的研究结果还表明:该算法能够发现一些其他基于模型的聚类算法可能遗漏的聚类结构。
1 引言
如今,随着高通量测序技术的发展,研究人员可以进行越来越复杂的转录组动态研究。这类研究通常通过对反转录后的RNA分子进行测序来实现,称为RNA测序(RNA-seq)。对RNA-seq数据的研究有助于深入理解转录活性的变化如何反映不同的细胞类型,并对表型差异产生影响。
从RNA-seq数据中获得新见解的一种方式是识别共表达基因的群体(即聚类)。这有助于研究人员锁定参与相似生物过程的基因,从而推动新药设计,或识别出潜在的共调控候选基因。识别共表达基因的聚类还可用于表征**孤儿基因(尚未知功能的基因)**的生物学功能。
目前,已有多种聚类算法被提出用于RNA-seq数据分析。然而,这类数据具有一些显著特征,使得其建模相当困难:
-
首先,它们往往呈现出高度偏态分布且动态范围广;
-
其次,基因长度与测序读数之间通常存在正相关关系;
-
第三,这些数据几乎总是呈现过度离散特性(即方差大于均值)。
多数基于模型的RNA-seq聚类方法,通常将每个聚类视为一种不同的分布,并将整个数据集建模为这些分布的有限混合体。这种方法的优势在于,它允许研究人员评估合适的聚类数量、聚类之间的距离,并对这些量进行假设检验。
现有文献中的大多数方法采用的是参数模型,即假设每个样本的数据分布属于某个特定分布族。所使用的分布类型包括泊松分布、负二项分布,甚至是为特定任务定制的复杂分布,如多元泊松-对数正态分布。
但在这种情况下,选择哪种分布往往是一项困难的任务,因为目前缺乏针对大多数多元离散分布的现成拟合优度检验方法。尽管参数方法具有计算成本低、解释性强的优点,但寻找一种非参数替代方案可能会更有益。
生物聚类任务的一个可行替代方法是使用非参数有限混合模型。与参数模型相比,非参数建模对数据本身的分布特性假设更少,更灵活。
更具体地说,我们建议将混合模型中的组成分布仅视为普通的概率密度函数,无需将其归类到某个特定分布族中。非参数多元混合建模与聚类在统计学中已有较长时间的研究历史,多个方法已被提出以拟合此类模型并揭示相关聚类结构。然而,这些方法在生物信息学领域的应用仍十分有限;据我们所知,它们从未被用于转录组数据的聚类任务。
在本研究中,我们建议使用一种可在RNA-seq数据上下文中拟合具有条件独立边缘分布的通用多元非参数混合模型的算法。该方法称为npMSL(非参数最大平滑似然)方法,最初在文献 [6] 中提出。
该方法对应的算法是一个MM(最大化-最小化)算法,具有类似EM算法的单调收敛性质,并且保证最终收敛。
本文接下来的内容安排如下:
-
在 方法部分,我们将详细描述所使用的模型,引入必要的符号,并定义执行聚类任务所用的算法;同时介绍如何选择聚类数量的模型选择方法;
-
接着是 模拟研究部分,我们使用一个合成数据集分析该方法的性能;
-
在 真实数据分析部分,我们详细介绍一个人类前列腺癌数据集,以展示我们的方法;
-
结果部分提供了基于可视化方法和GO功能富集分析的结果展示;
-
本研究表明,该算法有能力发现其他混合模型聚类方法可能忽略的、在实践中具有重要意义的聚类解决方案;
2 方法
设 Yij 表示第 i 个生物实体在第 j 个条件下的数字基因表达测量值(i=1,…,n,j=1,…,d),其观测值为 yij。于是数据 y 是一个 n×d的矩阵,记录了所有观测与变量的数字基因表达值。令 yi 表示观测 i 的 d-维向量。
2.1 条件独立测量的非参数混合模型
传统上,RNA-seq 数据聚类通常使用参数化混合模型。然而,据我们所知,尚未有人尝试将非参数方法用于 RNA-seq 数据的模型聚类。相比之下,这种方法已在其他领域(如发展心理学、水文学等)中得到应用。
该方法不假定簇的密度函数属于某个参数族(如高斯、泊松等)。具体地,数据被假定来自 m 个不同的簇,其中第 k 个簇的密度为 fk。进一步假设,每个密度函数 fk 可以表示为其边际密度的乘积:
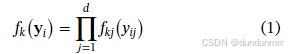
从而,任何观测 Yi的密度函数可用非参数有限混合模型表示为:
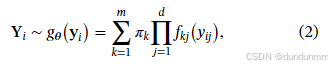
其中 π=(π1,…,πm)满足 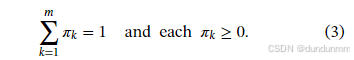 ,且每个 πk≥0。
,且每个 πk≥0。
上述建模假设了条件独立性,这在 RNA-seq 的参数混合建模中也常见。与传统模型不同,该方法不要求任何边际密度 fjk来自于特定的参数族,因此是一种实质性的泛化。
2.2 推理方法
定义 Zik∈{0,1}为伯努利变量,表示观测 i 来自于第 k 个组。整个数据由可观察和不可观察部分组成,因此适合使用 EM 类算法来估计模型 (2) 的参数。
我们采用了一种最小化模型 (2) 所生成数据的平滑对数似然函数的算法,即 npMSL(nonparametric Maximum Smoothed Likelihood)算法,该算法具有类似 EM 的单调性保证。
以下为算法步骤:
-
使用核密度函数 K(⋅),定义平滑操作:
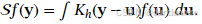
-
定义混合操作
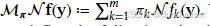 ,则:
,则:E 步:
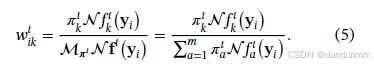
M 步:
-
更新混合权重:
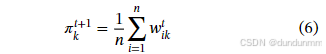
-
更新密度估计:
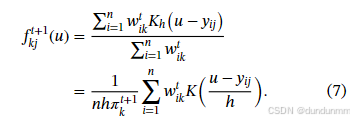
-
该算法可推广到坐标块建模,即假设某些条件具有相同分布。在这种情况下,只需在 M 步中对密度估计步骤作出调整。
2.3 带宽与核函数的选择
核函数选择对估计结果影响较小,但带宽选择对非参数混合建模具有重大影响。我们采用 Silverman 经验法则:
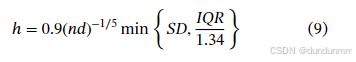
其中 SD 为标准差,IQR 为四分位差。尽管粗糙,但该方法适用于实际中简化模型设置,避免打破算法单调性。
2.4 聚类数目的选择
npMSL 算法要求预先设定聚类数,但现实中聚类数常不可知。与参数模型不同,非参数模型缺乏有效手段来确定聚类数。
我们采用基于 高斯混合模型与熵聚类 的间接方法作为参考基线,即通过 mclust 包进行建模、BIC 优化与熵驱动聚类组合。该方法虽依赖高斯假设,但能提供非参数建模中的可比较参考。
实验
模拟研究
作为第一步,我们进行了一项模拟研究,将我们提出的方法与三种其他基于模型的聚类方法进行了比较:文献[1]中的泊松模型方法、文献[9]中的变换方法以及文献[17]中的组合方法。为了避免模拟数据“量身定制”某个特定分布的问题,我们采用了文献[2]中建议的、基于负二项分布生成的一组数据集。
我们将在此仅简要介绍数据生成方案,详细内容可参考文献[2]。
首先,设 Ngij 表示在处理组 i、重复 j下,映射到基因 g 的读取计数(g=1,…,G,i=1,…,I,j=1,…,ni),其中 G 为总基因数,I 为处理组数,ni为每个处理组的重复数。因此,数据由计数 Ngij 组成。
其均值设为 λgij,满足以下模型:
![]()
其中:
-
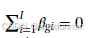 ;
; -
sgij是偏移量;
-
αg 是基因 g 在所有处理中的几何平均表达水平;
-
βgi则表示处理组 i 中相对于平均表达水平的偏差。
为建模超离散性,计数的方差设为:
![]()
其中 ϕg是基因 g 的离散参数。
聚类性能评估
我们通过比较不同方法生成的聚类与真实聚类(由 Z0={Zg0}定义)之间的一致性来评估聚类性能。采用两种常用指标:
-
对偶灵敏度(pairwise sensitivity):正确聚类在一起的基因对所占比例。
-
对偶特异性(pairwise specificity):正确划分到不同簇的基因对所占比例。
这两个指标在文献[2,20,21]中被广泛使用。
我们的方法(npMSL)与另外三种方法在以下两种情形下进行了比较:
-
(图1A)真实簇数 K=7已知;
-
(图1B)簇数设为错误的 K=10K,用于模拟真实应用中簇数未知的情形。
注意:簇数设定只对 npMSL、泊松方法、变换方法有影响,对组合方法无影响(其能自动确定簇数)。
实验中,设表达波动参数 ηε从 0.2 增至 2,步长为 0.25,ημ,ηα,ηϕ\eta_\mu, \eta_\alpha, \eta_\phi 固定为 1。每组参数组合下生成 100 个数据集,分别使用四种方法聚类,并采用上述两项指标进行评估。
npMSL 方法中,带宽选用 Silverman 经验法则(公式9),使用高斯核函数进行平滑。
所有指标在 100 次模拟中以平均值 ± 90% 置信带的形式展示。泊松与变换方法的置信带极窄,在图中难以察觉。图1A 和 1B 中称组合方法为 mclust 方法。
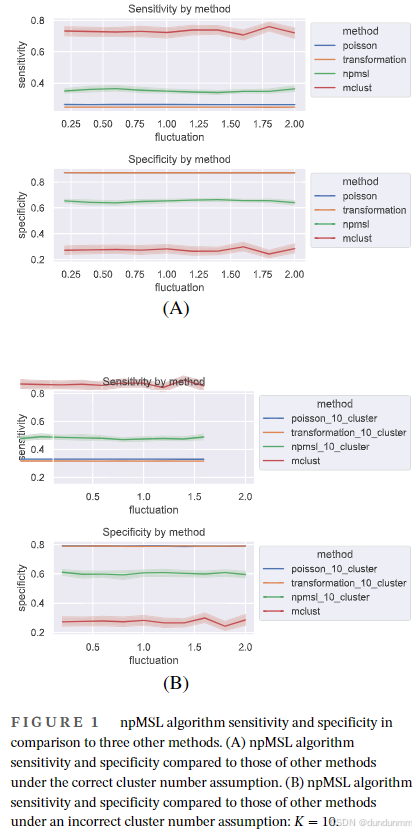
实验结果总结
在真实簇数设定正确的情况下:
-
npMSL 方法的灵敏度明显优于泊松与变换方法,但低于组合方法;
-
特异性方面,泊松与变换方法几乎相同,npMSL 略差,组合方法最差。
在簇数设定错误时,趋势类似:
-
npMSL 的灵敏度仍优于泊松与变换方法,低于组合方法;
-
但其特异性仍弱于前两者,优于组合方法。