DoubletFinder去除双细胞分析学习
在单细胞RNA测序过程中,有时两个或多个细胞可能在制备过程中意外结合成一个单一的"假细胞",称为双峰细胞或双倍体。这些双峰细胞可能会扭曲数据分析和解释,因此,需要使用一些方法对它们进行识别和剔除。其中DoubletFinder是最常用的一个工具。
官方对DoubletFinder输入的对象和参数介绍

-
seu:这是一个完全处理过的 Seurat 对象,即已经完成了数据规范化(NormalizeData)、寻找变异基因(FindVariableGenes)、数据标准化(ScaleData)、主成分分析(RunPCA)和 t-SNE 分析(RunTSNE)。
-
PCs:指定用于分析的统计显著的主成分数量,例如 PCs = 1:10。
-
pN:定义生成的人工双倍体数量,以合并的真实-人工数据比例表示。默认设置为 25%,根据 McGinnis, Murrow 和 Gartner 在 2019 年的 Cell Systems 文章,DoubletFinder 的表现在很大程度上与 pN 参数无关。
-
pK:定义用于计算 pANN 的 PC 邻域大小,同样以合并的真实-人工数据比例表示。没有默认值,因为每个单细胞 RNA 测序数据集都应该调整 pK 值。最优的 pK 值应该使用下面描述的策略来估计。
-
nExp:定义用于做出最终双倍体/单倍体预测的 pANN 阈值。这个值最好从 10X 或 Drop-Seq 设备的细胞加载密度中估计,并根据同源双倍体的预估比例进行调整。
官网文档中对示例数据的要求和参数进行了解释。其中seu对象是建议提前进行处理的。PC值其实可以按照使用者降维聚类选择的值而定。pN就默认25%即可。pK和nExp有函数可以进行计算。
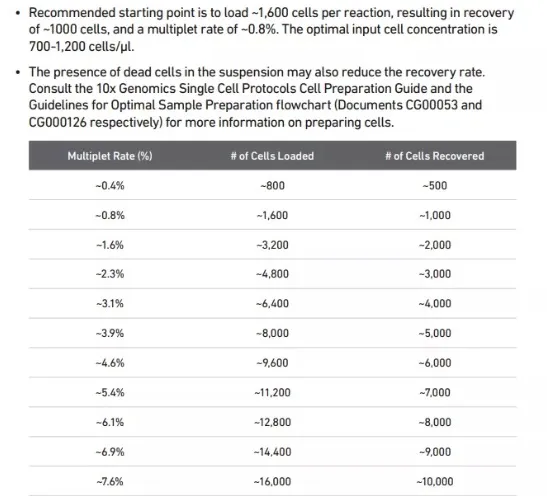
下面的表格是DoubletRate参数选择的参考文件(10X),在分析之前参照这个表格上边的细胞数选择DoubletRate值。
步骤流程
1、导入
scRNA是多样本已经合并完成并进行过标准流程后的数据集
rm(list=ls())
library(DoubletFinder)
library(BiocParallel)
library(qs)
library(Seurat)register(MulticoreParam(workers = 4, progressbar = TRUE))
scRNA <- qread("./sce.qs")
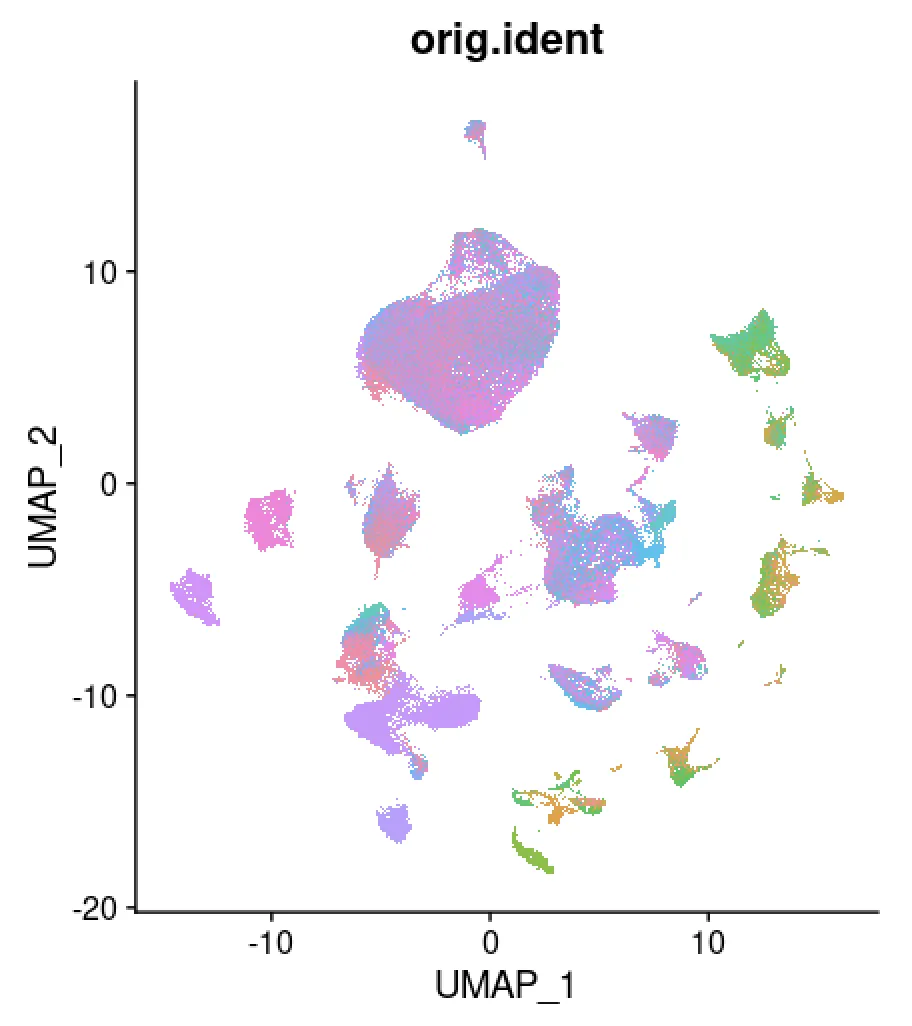
table(scRNA$orig.ident)# check一下
DimPlot(scRNA,pt.size = 0.8,group.by = "orig.ident",label = F)
2、DoubletFinder分析
一般是建议按照每个cluster进行分析,SCT参数是指SCTransform,如果是其他方式比如harmony之后的,可以考虑不选择T。
#单个分开,用来做DoubletFinder
sce_list <- SplitObject(scRNA, split.by = "orig.ident")pc.num <- 1:30
DoubletRate = 0.023 # 大约4800的细胞
# 找到pK
sweep.res <- paramSweep(sce_list[["C1"]], PCs = pc.num, sct = F) # sct也可以选择T
sweep.stats <- summarizeSweep(sweep.res, GT = FALSE)
bcmvn <- find.pK(sweep.stats)
pK_bcmvn <- bcmvn$pK[which.max(bcmvn$BCmetric)] %>% as.character() %>% as.numeric()# 计算homotypic doublets的比例和预期的doublet数目
homotypic.prop <- modelHomotypic(sce_list[["C1"]]$seurat_clusters) # 最好提供celltype
nExp_poi <- round(DoubletRate * ncol(sce_list[["C1"]]))
nExp_poi.adj <- round(nExp_poi * (1 - homotypic.prop))# 使用确定的参数鉴定doublets
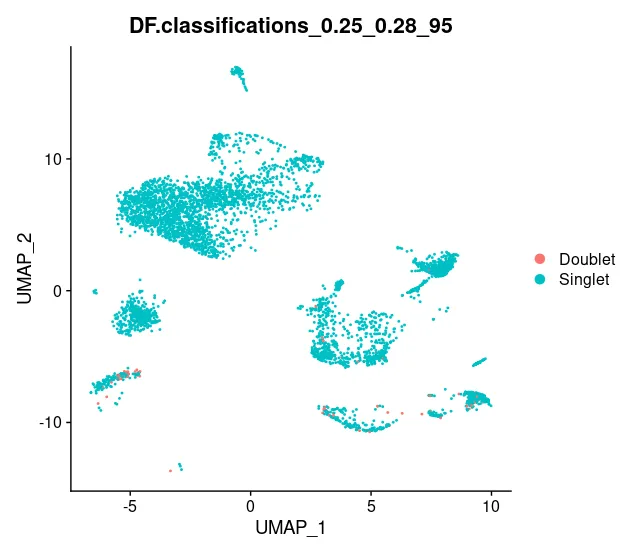
sce_list[["C1"]] <- doubletFinder(sce_list[["C1"]], PCs = pc.num, pN = 0.25, pK = pK_bcmvn, nExp = nExp_poi.adj, reuse.pANN = F, sct = F) # 也可以选择T# 图片展示
DimPlot(sce_list[["C1"]], reduction = "umap", group.by = "DF.classifications_0.25_0.28_95")
对sce_list中的每一个样本都需要走一遍流程,之后再进行合并。
流程不复杂,C1名称需要按照自己数据修改,如果样本量多的话步骤会比较繁琐,使用者可考虑进行函数封装。
同时也有一些观点认为应谨慎处理双细胞,因为这些双细胞毕竟是人为定义的,那么是不是真的是双细胞其实也是要思考的,所以可以先进行双细胞的检测不删除,等后续观察细胞分群的情况以及功能富集等一些操作之后再做考虑。
参考资料:
1、DoubletFinder: https://github.com/chris-mcginnis-ucsf/DoubletFinder
2、单细胞天地:https://mp.weixin.qq.com/s/O0U8vlMIG9vUVE3FK08LJg
致谢:感谢曾老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -